


Rythmologie et rythmo interventionnelle
Publié le 23 juin 2015Lecture 8 min
La dysplasie/cardiomyopathie ventriculaire droite arythmogène
P. CHARRON, Centre de référence pour les maladies cardiaques héréditaires, CHU Pitié-Salpêtrière, Paris
Un peu d’histoire
Nous devons probablement à Giovanni Maria Lancisi, professeur d’anatomie à Rome, la première description de la maladie en 1736 dans son ouvrage « De motu cordis et aneurysmatibus ». Ignorée pendant plus de deux siècles, la maladie a été redécouverte et décrite sous le nom de dysplasie ventriculaire droite arythmogène dans les années 1970. La description moderne de la maladie a été rapportée en 1982 par Frank Marcus et Guy Fontaine(1), à travers les cas de 24 patients adultes dont la vaste majorité se présentait avec une tachycardie ventriculaire récidivante à type de retard gauche. Cet article princeps décrivait les autres caractéristiques de la maladie : les anomalies ECG, dominées par les ondes T négatives de V1 à V4 et des ondes (epsilon) de post-excitation, les anomalies morphologiques du ventricule droit, avec la localisation préférentielle au niveau du « triangle de la dysplasie » (infundibulum, apex, segment diaphragmatique), les anomalies histologiques avec des fibres myocardiques raréfiées et remplacées par un tissu adipeux et fibreux.
Le terme de « dysplasie », utilisé initialement, qualifiait un trouble de développement possiblement embryonnaire, par analogie avec une forme extrême de maladie ventriculaire droite à révélation volontiers néonatale, la maladie de Uhl (décrite par Henry Uhl en 1952). Le concept physiopathologique a ensuite évolué, et le terme de « cardiomyopathie » ventriculaire droite a progressivement remplacé celui de « dysplasie », pour désigner la fréquente apparition retardée, progressive avec l’âge(2,3).
C’est cependant à la génétique moléculaire que l’on doit d’avoir franchi une étape majeure dans la compréhension de la maladie. Un premier gène muté, codant la plakoglobine, a tout d’abord été identifié par criblage du génome en 2000 par l’équipe de William J. McKenna dans une forme très rare appelée maladie de Naxos, associant atteinte cardiaque et cutanée (cheveux laineux et hyperkératose palmo-plantaire). Ensuite, des mutations responsables des formes communes de dysplasie/cardiomyopathie ventriculaire droite (CVDA/DVDA) ont été identifiées dans d’autres gènes codant pour les protéines du desmosome(4), au premier chef la plakophiline-2. Le travail visant à comprendre le mécanisme des mutations se poursuit. L’irruption du test génétique dans la pratique clinique a, quant à elle, déjà débuté(5).
Le cas de la famille K...
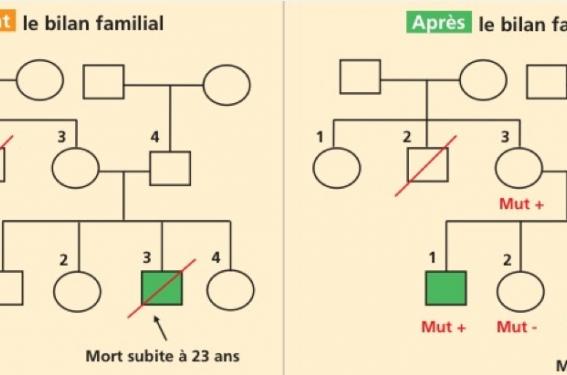
M. et Mme K... sont vus en consultation de cardio-génétique à la suite du décès par mort subite de leur fils, âgé de 23 ans (voir l’arbre généalogique, figure 1, individu III-3). Il n’avait aucune maladie cardiaque connue auparavant. C’est l’autopsie pratiquée à visée diagnostique qui a permis de poser le diagnostic de CVDA/DVDA.
Arbre généalogique de la famille K... (avant et après le bilan familial)
Figure 1. Les générations I à III sont représentées. Symbole plein : atteint de DVDA; symbole vide : absence de DVDA ; symbole hachuré : borderline ; symbole carré : homme; symbole rond : femme. Mut+ et Mut- : porteur et non-porteur de la mutation.
L’histoire familiale ne retrouve pas d’autre cas de dysplasie ou de cardiomyopathie avérée, et les apparentés proches (fratrie, parents) n’ont pas de symptomatologie, en dehors de quelques palpitations chez un frère (III-1), n’ayant pas conduit à un bilan cardiaque. On note également dans la branche maternelle une mort subite à l’âge de 57 ans, sans maladie documentée.
Les parents se posent de nombreuses questions sur la maladie, ses aspects génétiques et surtout sur le risque potentiel chez leurs trois autres enfants (III-1, III-2 et III-4). Un bilan cardiologique systématique est alors organisé au sein de la famille. En parallèle, du tissu autopsique est transféré dans le laboratoire de notre hôpital pour analyse génétique post-mortem.
Généralités sur les aspects génétiques de la maladie
La prévalence de la CVDA/DVDA dans la population générale est estimée entre 1 sur 2 000 et 1 sur 5 000(3) (figure 2). Les formes familiales ont longtemps été sous-estimées, mais des études systématiques ont montré qu’elles constituaient 30 à 50 % des cas. Le mode de transmission est autosomique dominant avec, par conséquent, un risque de transmission de 50 % à chaque apparenté au 1er degré. Une exception notable est une forme rare, la maladie de Naxos, dont la transmission est autosomique récessive. Des mutations nombreuses ont été identifiées ces toutes dernières années(4), essentiellement dans 5 gènes codant pour des protéines du desmosome : la plakophiline-2 (PKP2), la desmogléine-2 (DSG2), la desmoplakine (DSP), la desmocolline-2 (DSC2), la plakoglobine (JUP). Les conséquences précises des mutations ne sont pas connues, mais l’hypothèse principale est que l’altération des desmosomes (jonctions intercellulaires) induite par les mutations entraîne une dégénération progressive des cardiomyocytes avec mort cellulaire, suivie d’une réparation par remplacement fibro-adipeux.
Dans une étude collaborative française analysant 135 patients indépendants, une mutation a été identifiée chez environ 50 % des patients(6), que le contexte soit celui d’une forme familiale ou celui d’une forme sporadique. Dans ce même travail, la répartition des gènes cités plus haut était respectivement de 31,5 %, 10 %, 4,5 %, 1,5 %, 0 %. Quelques rares mutations ont été identifiées dans d’autres gènes (récepteur à la ryanodine, TGF-bêta 3, protéine TMEM43, desmine).
Plusieurs études ont observé que la maladie était caractérisée par une pénétrance incomplète des mutations (probabilité de développer la maladie cardiaque < 100 %, en particulier chez les femmes) et une grande variabilité d’expression clinique (coexistence dans la même famille de forme précoce/sévère et de forme tardive/non compliquée). Il est trop tôt pour dire si certains gènes ont une valeur pronostique péjorative, mais des données préliminaires suggèrent que le pronostic peut être modulé selon le gène sous-jacent (pronostic plus sévère en présence de mutation du gène DSP ou DSG2) ou la présence de mutations multiples (4 % des patients dans l’étude française)(6).
Figure 2. Les principales manifestations de la CVDA/DVDA (d’après(3)).
Prise en charge clinico-génétique dans la famille K...
La prise en charge de la famille K... illustre l’intégration des connaissances génétiques au service d’une amélioration de la prise en charge médicale de la famille. La prise en charge a débuté dans cette famille par la consultation de conseil génétique vis-à-vis des parents, après la mort de leur enfant et la découverte de la maladie à l’autopsie.
La consultation a été l’occasion de donner aux familles une information sur les aspects génétiques de la maladie, son mode de transmission (donc l’identification des apparentés à risque), son histoire naturelle, son expressivité clinique variable. La consultation de génétique a permis également de discuter la réalisation d’une analyse génétique moléculaire, à partir du tissu autopsique de leur enfant, l’identification potentielle d’une mutation pouvant s’avérer utile pour guider la surveillance cardiologique au sein de la famille.
Enquête cardiologique familiale et prévention de la mort subite
Du fait de la très grande fréquence des formes génétiques et/ou familiales (toute CVDA/DVDA relève probablement d’une origine génétique), de la gravité potentielle de la maladie (le risque de mort subite par troubles du rythme ventriculaire est d’environ 1 à 2 % par an) et du bénéfice d’un traitement précoce (intérêt du traitement bêtabloquant notamment, d’une restriction d’activité sportive), il est souhaitable de préconiser un bilan cardiaque familial systématique, même devant un cas en apparence sporadique de la maladie. Cette attitude a été récemment préconisée par le groupe de travail « Cardiomyopathies » de la Société européenne de cardiologie(5) et les modalités en ont été précisées (le bilan s’adresse à tous les apparentés au 1er degré d’un patient atteint de CVDA/DVDA, au moins à partir de l’âge de 10 ans, avec ECG, échographie, ECG-HA, Holter-ECG, et poursuite d’une surveillance même si le 1er bilan est normal).
La famille décrite ici illustre l’intérêt d’un bilan cardiologique systématique, car il a permis de dépister une expression cardiaque très probable de la maladie (ondes T négatives en précordial, présence de potentiels tardifs, anomalie segmentaire en échographie) chez un frère paucisymptomatique (quelques palpitations, sujet III-1), et une expression possible de la maladie (ESV isolées abondantes) chez une sœur (III-4). Le bilan cardiologique a été approfondi avec IRM cardiaque et épreuve d’effort. Un traitement bêtabloquant a ensuite été proposé, de même qu’une restriction d’activité sportive.
Diagnostic génétique post-mortem et test prédictif chez les apparentés
Parallèlement au bilan cardiaque familial a été organisée une analyse génétique à partir de tissu congelé conservé par le service d’anatomie pathologique ayant pratiqué l’autopsie. Cette procédure de test génétique post-mortem, ou « autopsie moléculaire », se développe actuellement de plus en plus. L’analyse génétique moléculaire a permis d’identifier ici une mutation hétérozygote du gène PKP2 (plakophiline-2). La mutation aboutit à un codon-stop direct. Ce résultat confirme l’origine génétique et le mode de transmission autosomique dominant. Surtout il permet de disposer d’un outil diagnostique supplémentaire pour les investigations chez les apparentés, en accord avec la révision récente des critères diagnostiques internationaux de CVDA/DVDA(2) qui considère l’identification d’une mutation comme un critère « majeur » (le diagnostic étant porté en présence de 2 critères majeurs ou de 1 critère majeur et de 2 mineurs ou bien de 4 critères mineurs issus de différentes catégories).
Dans la famille considérée ici, une sœur (III-4) et un frère (III-1) se sont révélés porteurs de la mutation, et cela a permis d’interpréter les anomalies cardiaques mineures de la sœur comme relevant d’une forme débutante de la maladie. De plus, le test génétique prédictif a permis d’identifier la mutation chez la mère (II-3) du patient décédé, alors que son propre bilan cardiaque est revenu normal. Cela illustre un cas de pénétrance incomplète avec un parent transmetteur n’ayant pas développé la maladie (fréquemment observé dans cette maladie), qui permet d’orienter la surveillance dans une branche précise de la famille. À l’inverse, le prélèvement sanguin réalisé chez le père du patient décédé et chez l’autre sœur du patient a montré l’absence de la mutation. Il n’y a donc pas de surveillance cardiologique à organiser pour eux, et aucun bilan génétique ou cardiaque à prévoir dans la branche familiale paternelle.
Ce qu’il faut retenir
La cardiomyopathie/dysplasie ventriculaire droite arythmogène (CVDA/DVDA) est une maladie génétique à transmission autosomique dominante, et une mutation peut être identifiée chez environ 50 % des patients/familles dans l’un des gènes codant pour les protéines du desmosome.
La dimension familiale et génétique de la CVDA/DVDA doit être prise en compte et conduire dans tous les cas à une surveillance cardiologique chez les apparentés, y compris à l’âge adulte, de façon à permettre un diagnostic et une prise en charge précoces.
Il est également possible de proposer la réalisation d’un test génétique, de façon à optimiser la prise en charge des patients et de leurs apparentés, notamment dans une optique diagnostique ou prédictive.
Pour plus d’information, consulter le site internet du centre de référence pour les maladies cardiaques héréditaires : http://www.cardiogen.aphp.fr
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :

Articles sur le même thème
publicité
publicité