

Rythmologie et rythmo interventionnelle
Publié le 25 jan 2015Lecture 10 min
Le syndrome du QT long congénital
I. DENJOY, J.-M. LUPOGLAZOFF, Centre de référence des maladies cardiaques héréditaires, CHU Lariboisière et Robert-Debré, Paris
Le syndrome du QT long congénital (SQTL) est caractérisé par une anomalie de la repolarisation ventriculaire sur l’ECG de surface (QTc > 440 ms), associée à un risque significatif de syncope ou de mort subite du fait de troubles du rythme ventriculaire à type de torsades de pointes ou de fibrillation ventriculaire. L’exercice physique ou le stress émotionnel sont des facteurs déclenchants fréquents de ces syncopes ou morts subites. Le caractère familial de ce syndrome a été noté depuis de nombreuses années, et l’origine génétique en a été confirmée avec l’identification dès 1992 du premier gène muté associé à ce syndrome (gène KCNQ1). Depuis, 12 gènes ont été identifiés avec plusieurs centaines de mutations connues et répertoriées chez les sujets atteints. La majorité des anomalies génétiques concerne trois gènes principaux impliqués dans la repolarisation ventriculaire et permet de catégoriser le SQTL en plusieurs sous-types, dont les plus fréquents sont les formes LQT1, LQT2 et LQT3. La plupart des familles ont leur mutation propre, ce qui fait parler de mutation privée. Certaines caractéristiques cliniques, telles que la morphologie de l’onde T, la réponse de l’intervalle QT lors de l’effort, les facteurs déclenchants des troubles du rythme et la réponse au traitement, peuvent être influencées par le gène muté. La gravité de ce syndrome tient aux syncopes qui sont dues à des troubles du rythme ventriculaire polymorphes, rapides, typiquement des torsades de pointes, et au risque de mort subite par fibrillation ventriculaire. Les avancées moléculaires depuis 1992 ont ouvert la voie à de nombreux travaux qui permettent d’intégrer le test génétique dans la prise en charge de la maladie.
Épidémiologie
L’incidence des mutations est au moins de 1 pour 2 000 personnes. Cette estimation a été établie à partir des données obtenues au cours des bilans familiaux et en tenant compte de l’incidence des patients avec des mutations hétérozygotes composites.
Les différentes formes de SQTL
Le LQT1 est la forme la plus fréquente de SQTL. Il est dû à des mutations dans KCNQ1 entraînant une perte de fonction du canal potassique qui sous-tend le courant Iks. Dans cette forme, les syncopes ou morts subites surviennent au cours d’une émotion ou d’un effort physique tel que la natation par exemple. Dans quelques rares cas, les patients héritent de chacun de leurs parents d’une mutation dans KCNQ1 et sont atteints d’une forme particulièrement sévère associant un allongement de l’intervalle QT très important, une surdité congénitale bilatérale et un risque majeur de mort subite (syndrome de Jervell et Lange-Nielsen).
La forme LQT2 est due à une mutation dans le gène KCNH2 qui code pour un autre canal potassique qui sous-tend le courant potassique Ikr. Dans cette forme, les syncopes ou morts subites peuvent survenir lors de stress ou au repos. Les facteurs déclenchants sont parfois des stimulations sonores importantes telles que la sonnerie du téléphone, le réveil. Sur l’ECG, l’onde T est caractérisée par un aspect en double bosse.
La forme LQT3 est due à des mutations dans le gène du canal sodique cardiaque SCN5A. L’aspect électrocardiographique se caractérise par un QT particulièrement long, une onde T tardive et pointue et un risque très élevé de mort subite par fibrillation ventriculaire.
Le diagnostic
Les symptômes les plus fréquents du syndrome du QT long congénital sont les syncopes et l’arrêt cardiaque . Le diagnostic peut être posé à l’occasion de bilans familiaux chez des individus asymptomatiques, mais avec un intervalle QTc > 440 ms sur l’ECG.
Le diagnostic différentiel inclut toutes les causes de syncope chez le sujet jeune : syncope vaso-vagale, cardiomyopathie hypertrophique, voire tachycardie ventriculaire catécholergique. L’examen clinique est normal, et l’échocardiographie ne retrouve aucune anomalie morphologique chez ces patients.
L’ECG est l’examen clé de ce diagnostic. Il montre un intervalle QTc allongé (> 440 ms) associé à des anomalies de morphologie, variables selon les génotypes en cause. Il peut être parfois difficile d’établir avec certitude le diagnostic sur le simple électrocardiogramme. Un enregistrement Holter peut faciliter le diagnostic par la mesure de la durée de l’intervalle QT sur des périodes de rythme cardiaque stable à 60/min et par la mise en évidence d’une anomalie morphologique de l’onde T.
Le test d’effort peut être utile afin de documenter la non-adaptation de l’intervalle QT aux variations de fréquence, mais il ne permet pas de déclencher les troubles du rythme responsables des syncopes.
Le recueil de l’histoire familiale est essentiel, à la recherche de syncopes, d’antécédents de mort subite, de noyade, de mort subite inexpliquée du nourrisson.
Test génétique
Le diagnostic du syndrome du QT long congénital reste avant tout clinique, et la mise en évidence d’une mutation chez les sujets atteints permet de le confirmer. Chez les patients symptomatiques ayant des anomalies spécifiques de l’onde T, il est possible de prédire le gène en cause dans 70 à 80 % des cas. Des mutations dans les trois principaux gènes chez les sujets atteints sont retrouvées dans 50 % des cas en moyenne. L’absence de mutation détectée chez les patients restants est vraisemblablement due à des difficultés techniques (variant non codant) ou à des gènes responsables du SQTL non encore identifiés. Un test génétique négatif chez un patient cliniquement atteint ne remet pas en cause le diagnostic.
En pratique, le test génétique est utile dans deux situations :
- premièrement, en cas de diagnostic clinique fortement suspecté, car la forme génétique peut influer sur les choix thérapeutiques et sur la stratification des risques ;
- deuxièmement, lorsque la mutation familiale est connue car elle peut être recherchée chez tous les collatéraux, ascendants et descendants de cette famille. Il s’agit alors d’un diagnostic présymptomatique.
Stratification du risque
Le paramètre le mieux corrélé au risque de syncope et de mort subite dans le syndrome du QT long congénital est la durée de l’intervalle QTc. L’analyse de 647 patients LQT1, LQT2 et LQT3 a permis d’établir l’incidence des syncopes et des morts subites avant l’âge de 40 ans à 20 % si l’intervalle QTc est inférieur à 450 ms et à plus de 70 % chez les patients dont l’intervalle QTc, en l’absence de traitement, est supérieur à 498 ms.
Traitement
L’absence d’essai randomisé dans le SQTL reflète la relative rareté de cette maladie et l’hétérogénéité du type et de la sévérité des présentations cliniques. Les données permettant d’établir une prise en charge sont généralement issues de registres larges et de centres de référence. Les sujets à très faible risque de mort subite (patients âgés, asymptomatiques avec un intervalle QTc normal, diagnostiqués simplement à l’occasion du bilan génétique) peuvent probablement ne pas être traités. Il faut simplement rappeler chez ces sujets la contre-indication formelle de médicaments allongeant la repolarisation et susceptibles de favoriser des torsades de pointes.
La très grande majorité des patients avec SQTL relève d’un traitement bêtabloquant et éventuellement d’un défibrillateur automatique implantable (DAI).
Les bêtabloquants
La pierre angulaire du traitement du syndrome du QT long congénital est le traitement bêtabloquant. Des traitements bêtabloquants ayant une longue durée de vie, tels que le nadolol, sont habituellement utilisés. La posologie et l’effet de ces traitements sont évalués par la réalisation d’enregistrements Holter avec pratique d’un exercice ou d’une épreuve d’effort permettant de vérifier la limitation des fréquences cardiaques à 130/min. On estime également qu’un patient est correctement « bêtabloqué » si la fréquence de repos diminue de 20 %. Les bêtabloquants ne raccourcissent pas le QT. Des études observationnelles réalisées avant et après la mise en route d’un traitement bêtabloquant montrent une survie plus importante chez les patients recevant cette classe thérapeutique, tout particulièrement chez ceux ayant une forme LQT1 par rapport aux sujets LQT2 ou LQT3.
Le défibrillateur automatique implantable
Les indications du DAI ont été précisées récemment. Les patients avec SQTL et mort subite récupérée sont considérés comme des indications de type 1, particulièrement si l’intervalle QTc est supérieur à 500 ms. Les patients présentant des récidives de syncope sous bêtabloquant représentent des indications de type IIA et les indications de type IIB concernent l’implantation d’un DAI pour prévention primaire chez les patients ayant un intervalle QTc > 500 ms, particulièrement pour les formes génétiques LQT2 et LQT3.
Les activités sportives
Toute activité sportive de compétition est contre-indiquée en cas de SQTL. Cette recommandation concerne également les patients asymptomatiques diagnostiqués sur l’électrocardiogramme ou au cours d’un bilan familial. Les pratiques sportives de loisirs peuvent être autorisées sous couvert d’un traitement bêtabloquant conformément aux recommandations publiées (tableau).
Tableau. Recommandations pour la prise en charge du syndrome du QT long congénital…
Cas de la famille M...
(figure 1)
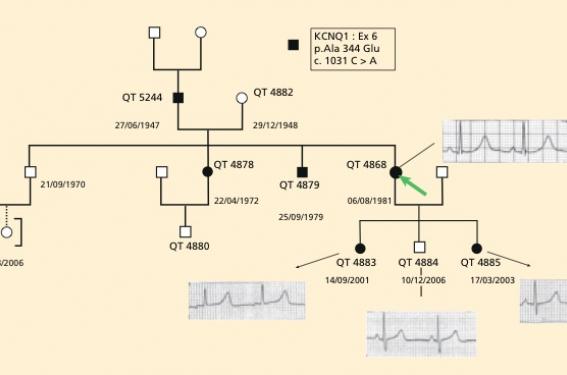
Mme M... est adressée par le Samu pour arrêt cardiocirculatoire récupéré, survenu à l’issue d’une syncope à la piscine. Il s’agit d’une femme de 28 ans en bonne santé, ayant 3 enfants, âgés respectivement de 8, 7 et 3 ans, asymptomatiques. D’après les témoins, Mme M... était au bord de la piscine, et s’est précipitée dans l’eau car elle a pensé qu’un de ses enfants s’était fait mal. Elle a présenté une perte de connaissance sous l’eau. Le premier tracé réalisé par les pompiers arrivés extrêmement rapidement montrait une fibrillation ventriculaire, choquée avec succès. L’électrocardiogramme à l’arrivée en soins intensifs montre (figure 1) un rythme sinusal à 80/min avec un intervalle QT allongé à 520 ms et une morphologie compatible avec une forme LQT1. À l’interrogatoire de la patiente, on apprend qu’elle a présenté des syncopes d’effort à 3 reprises à l’adolescence, alors qu’elle courait après l’autobus ou qu’elle était en train de faire du vélo.
Figure 1. Les cercles symbolisent les sujets de sexe féminin, les carrés les sujets de sexe masculin. Le symbole est noir lorsque la mutation trouvée chez le cas index (flèche) a été identifiée chez les apparentés. Les ECG montrent un QTc allongé chez le cas index (QTc 490 ms) et chez ses 2 filles (QTc 485 ms sujet QT 4883, et QTc 460 ms sujet QT 4885), alors que l’ECG de son fils est normal (QTc 410 ms sujet 4884).
L’histoire clinique et la morphologie de l’onde T sur l’ECG sont en faveur d’un syndrome du QT long congénital plutôt de type 1. Un traitement par nadolol 80 mg/j est instauré, et la liste des médicaments allongeant l’intervalle QT est remise à la patiente. Après recueil de son consentement, une prise de sang est réalisée pour étude de l’ADN et recherche d’une mutation dans l’un des gènes connus pour donner le syndrome du QT long congénital. Le gène KCNQ1 sera analysé en premier en raison des circonstances de survenue de la syncope et de la morphologie de l’onde T. Dans le même temps, ses 3 enfants sont vus en consultation, et pour 2 d’entre eux l’électrocardiogramme montre un intervalle QT allongé (figure 1). Un traitement bêtabloquant préventif est institué chez les 2 enfants dont l’ECG est anormal (nadolol 50 mg/m2/j en 2 prises). L’enquête familiale élargie retrouve des antécédents de syncopes à l’effort à l’adolescence chez le père de Mme M... avec un intervalle QT allongé (QTC : 470 ms) sur son électrocardiogramme et un intervalle QT allongé chez un frère (QTc : 480 ms) et une sœur (QTc : 475 ms) actuellement asymptomatiques (figure 1).
Les Holter réalisés confirment la permanence de l’allongement de l’intervalle QTc. Un traitement bêtabloquant est institué chez le père puisqu’il a présenté des syncopes et chez tous les membres de la famille ayant une anomalie de l’électrocardiogramme. Par ailleurs, des prélèvements sanguins à visée génétique sont réalisés à tous les membres de la famille. Quelques mois plus tard, une mutation dans le gène KCNQ1 est identifiée chez Mme M... également retrouvée chez tous les sujets qui présentaient un intervalle QTc allongé à l’électrocardiogramme, confirmant le diagnostic de ce syndrome chez plusieurs membres de cette famille.
Patientes avec désir de grossesse
La sœur de Mme M..., dont le syndrome (QTc allongé et présence de la mutation familiale) a été diagnostiqué au cours de l’enquête familiale et qui est parfaitement asymptomatique, désire un enfant. La grossesse n’augmente pas particulièrement le risque de complication rythmique. En revanche, il est impératif de poursuivre le traitement bêtabloquant tout au long de la grossesse. C’est plutôt la période du post-partum étendu (9 mois), qui présente une augmentation significative du risque de complication rythmique. Il est donc impératif d’informer les femmes atteintes du syndrome de QT long congénital qu’elles ne doivent pas arrêter leur traitement bêtabloquant pendant et après la grossesse. Il existe également un risque de 50 % de transmission du syndrome. Les complications rythmiques fœtales sont exceptionnelles et se retrouvent essentiellement, lorsqu’elles existent, dans les formes LQT2 et LQT3. Il est néanmoins recommandé que les grossesses se déroulent dans des maternités de niveau III. En effet, la prise de bêtabloquants durant la grossesse est souvent associée à des petits retards de croissance in utéro et à des hypoglycémies néonatales. Lors de la naissance de l’enfant, avec une probabilité de 50 % d’avoir hérité de la mutation maternelle, seront réalisés un électrocardiogramme et une prise de sang à la recherche de la mutation familiale. En effet, dans la mesure où les complications de ce syndrome peuvent exister dès les premières heures de vie, il est important de connaître le statut atteint ou sain de l’enfant, ce qui permettra de mettre en route un traitement préventif des complications cardiaques.
Diagnostic prédictif chez les apparentés
Au cours du bilan familial initial, avant que la mutation familiale ne soit disponible, un simple électrocardiogramme et l’interrogatoire permettent de faire le diagnostic de syndrome du QT long congénital chez les apparentés. Lorsque la mutation familiale est identifiée, il peut être recherché directement chez tous les membres de la famille. Il existe, en effet, dans ce syndrome 5 % d’individus avec des valeurs d’intervalle QTc à la limite supérieure de la normale et néanmoins porteurs de l’anomalie génétique. Ces sujets nécessitent une prise en charge spécifique, voire un traitement médical par bêtabloquant.
Ce qu’il faut retenir
Le caractère familial et génétique du syndrome du QT long congénital doit être pris en charge et conduit dans tous les cas à une consultation cardiologique spécialisée pour tous les membres de la famille, y compris chez les enfants de façon à permettre de porter un diagnostic et d’avoir une prise en charge précoce et adaptée. Le test génétique permet d’améliorer la prise en charge des patients et de leurs apparentés dans diverses situations : test prédictif, test diagnostique, test pronostique. Les différentes dimensions de l’impact d’un test génétique, en particulier l’aspect psychologique, social et médico-légal, doivent être reconnues et prises en compte, souvent dans le cadre d’une approche multidisciplinaire.
La mise en place récente par le ministère de la Santé de centres nationaux de référence et régionaux de compétence pour les maladies rythmiques héréditaires devrait permettre d’améliorer la prise en charge cardiologique et génétique de cette pathologie : http://www.cardiogen.aphp.fr
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :

Articles sur le même thème
publicité
publicité