

Publié le 21 juin 2005Lecture 6 min
Remodelage ventriculaire
D. LOGEART, hôpital Beaujon, Clichy

Une des avancées majeures dans la compréhension de la physiopathologie de l'insuffisance cardiaque chronique a été la compréhension du processus de remodelage du ventricule gauche. Le remodelage est un terme générique désignant une série de modifications structurelles d'un tissu ou d'un organe, survenant au cours du temps et à des divers niveaux : anatomiques, histologiques et biologiques. En ce qui concerne le ventricule gauche, tout dommage ou modification durable des conditions de travail (infarctus, hypertension, fuite valvulaire, etc.) provoque un remodelage de ce ventricule. L'exemple clinique le plus classique est la dilatation du postinfarctus.
Si l'existence d'une dilatation et/ou d'une hypertrophie des cœurs défaillants est connue depuis le XIXe siècle, l'insuffisance cardiaque fut d'abord considérée comme un désordre hémodynamique (baisse de la performance cardiaque, du débit et de la pression artérielle, rétention hydrosodée, etc). Les travaux pionniers sur la compréhension du processus de remodelage ne datent que de 25 ans, grâce notamment au modèle d'infarctus expérimental chez le rat. Plus grande était l'étendue de l'infarctus, plus importante était la dilatation du ventricule au cours du temps et plus importante était la mortalité. Ensuite, ces auteurs montrèrent qu'un traitement précoce par IEC pouvait freiner ce remodelage avec en parallèle une amélioration de la mortalité, contrairement aux traitements visant uniquement à améliorer l'hémodynamique. Ces résultats ont été suivis par de nombreux autres travaux expérimentaux et cliniques qui ont amené deux concepts majeurs pour notre pratique :
• le remodelage ventriculaire est associé à l'histoire naturelle de l'insuffisance cardiaque et notamment à son pronostic ;
• les traitements qui permettent de ralentir ou d'inverser ce processus de remodelage sont ceux qui ont un effet bénéfique sur le pronostic.
Physiopathologie du remodelage
Toute charge excessive de travail du cœur induit une hypertrophie — processus de « croissance » — des myocytes ventriculaires (une hyperplasie, ou multiplication des myocytes, semble aussi exister mais de façon marginale). En effet, l'ajustement de la masse cardiaque à la charge hémodynamique, c'est-à-dire aux contraintes s'exerçant sur les parois, est une caractéristique fondamentale (loi de Laplace : contraintes pariétales = volume x pression/masse). On parle d'ailleurs d'hypertrophie « compensatrice ».
Deux types d’hypertrophie
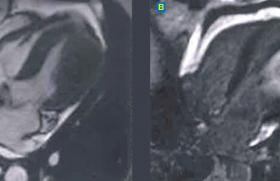
Sur le plan anatomique, on distingue deux types d'hypertrophie (figure 1) :
- l’hypertrophie concentrique, secondaire à l'hypertension artérielle ou au rétrécissement aortique, se caractérise par une augmentation de l'épaisseur des parois sans dilatation de la cavité, permettant une normalisation des contraintes pariétales systoliques et une augmentation de la force de contraction pour vaincre une postcharge élevée ;
- l'hypertrophie excentrique, observée dans les régurgitations valvulaires ou les cardiomyopathies dilatées primitives, se caractérise par une dilatation sans augmentation des épaisseurs pariétales, due à un glissement des couches de myocytes dans le grand axe ; cela permet une amélioration de la distensibilité et une réduction de l'augmentation de la pression diastolique provoquée par la surcharge de volume (la masse augmente aussi par hypertrophie des myocytes dans leur grand axe, permettant de réduire les contraintes diastoliques).
Dans le postinfarctus, il existe une hypertrophie du myocarde sain associée à une dilatation de la cavité.
Figure 1. Différentes formes anatomiques de remodelage.
Mécanismes du remodelage
Les mécanismes induisant ce remodelage sont complexes (figure 2). Toute une série de signaux permettent le transfert de l'information « augmentation des contraintes exercées sur le myocyte » à la réponse nucléaire « synthèse de nouvelles protéines ». Ces signaux débutent par une modulation de l'architecture du cytosquelette (intégrines, disques Z…) qui conduit à une activation nucléaire de la synthèse protéique. Entre les deux, se situe l'activation de différentes voies ou cascades de signalisation moléculaire, telles les voies de la calcineurine et des MAPK (mitogen-activated protein kinase). Il est intéressant de noter que l'assistance circulatoire, qui provoque une « décharge » majeure et prolongée du ventricule, induit un effet inverse sur le remodelage, ce qui confirme le rôle des contraintes mécaniques dans l'induction du processus.
Figure 2. Schématisation partielle du processus de remodelage au niveau cellulaire.
L'angiotensine II est largement impliquée dans cette signalisation. Sa sécrétion autocrine/paracrine augmente sous l'effet des contraintes mécaniques et les taux circulant augmentent par ailleurs quand s'est déjà installée une insuffisance cardiaque avérée ; en retour, elle stimule directement les voies de signalisation de la réponse hypertrophique. De même, les catécholamines, l'endothéline ou les cytokines (TNF), voient leur synthèse s'accroître avec l'évolution de la maladie et stimulent les processus cellulaires de remodelage.
Sur les plans histologique et cellulaire
Le remodelage s'accompagne de profonds remaniements résultant de l'activation de nombreux gènes. Au niveau des myocytes, sont observés un changement d'isoformes des protéines contractiles, une stimulation de la synthèse intracardiaque d'angiotensine II, des peptides natriurétiques, un allongement de la durée du potentiel d'action, une baisse du ratio mitochondrie/volume cellulaire… Ce processus touche également les autres composants cellulaires du myocarde ; il y a notamment une activation et/ou une prolifération des fibroblastes avec une synthèse accrue de collagène et le développement d'une fibrose interstitielle.
Au début du processus, on parle souvent d'hypertrophie « compensatrice » car l'épaississement des parois ventriculaires permet une normalisation des contraintes pariétales. C'est le cas notamment dans l'hypertension artérielle ou le rétrécissement aortique. Mais s'y associent rapidement des modifications biologiques, dont certaines sont à l'évidence néfastes, qui peuvent notamment induire une dysfonction diastolique. Finalement, c'est le déséquilibre entre effets bénéfiques et délétères qui conduit ou précipite l’évolution vers la phase dite « décompensée », qui correspond à l'insuffisance cardiaque. Il existe alors un véritable cercle vicieux entre les aspects péjoratifs du remodelage et la défaillance hémodynamique. Les modifications phénotypiques s'accentuent dans leur caractère délétère et concourent définitivement à l'aggravation de l'hémodynamique par dysfonction diastolique et systolique et à favoriser les arythmies : réduction de l'activité de repompage du calcium et surcharge calcique intracytosolique, désensibilisation des récepteurs adrénergiques, réduction du métabolisme oxydatif mitochondrial, activation de voies apoptotiques, fibrose et perte de distensibilité, etc. Cette transition vers l'insuffisance cardiaque chronique est plus ou moins rapide selon le type et la sévérité du dommage initial et/ou de la surcharge de travail cardiaque, mais probablement aussi en fonction de prédispositions génétiques modulant l'évolution du remodelage.
Remodelage, pronostic et traitement
Plusieurs études ont démontré que le diamètre télédiastolique est un facteur pronostique indépendant, notamment dans le postinfarctus. Dans l'hypertension artérielle, l'existence d'une hypertrophie ventriculaire est également un facteur indépendant de mauvais pronostic.
Les traitements agissant uniquement sur la composante hémodynamique de l'insuffisance cardiaque (inotropes positifs notamment) améliorent les symptômes mais sont inefficaces, voire délétères à terme sur la mortalité. À l'inverse, les agents bloquant les systèmes rénine-angiotensine-aldostérone (IEC, ARA II, spironolactone) et catécholergique (bêtabloqueurs) sont très efficaces.
La compréhension de la physiopathologie du remodelage a permis de situer la pertinence de ces médicaments qui bloquent des médiateurs cruciaux du remodelage ventriculaire. Dans l'étude SOLVD (Study Of Left Ventricular Dysfunction) qui incluait des patients avec FEVG ≤ 35 %, le volume télédiastolique VG a augmenté progressivement au cours des 3 mois de suivi (confirmant clairement l'existence d'un remodelage) ; le traitement IEC a réduit la mortalité et aussi précocement la dilatation, avec un effet se maintenant sur les 3 ans de suivi (figure 3). La même efficacité est observée avec les bêtabloqueurs, dont l'effet se s’ajoute à ceux des IEC.
Figure 3. A. Effets de l'énalapril sur le remodelage VG dans l'étude SOLVD. B. Effets du carvédilol dans l'étude Australia-New Zealand Heart Failure Research.
Quelles perspectives ?
Le développement de nouveaux agents thérapeutiques est largement axé sur le processus de remodelage. Les multiples voies de signalisation moléculaire de ce processus représentent autant de cibles potentielles bien que la plupart de ces voies soient redondantes et que le blocage de l'une d'entre elles risque d'être insuffisant. La thérapie cellulaire peut être incluse dans cette thérapie visant à amoindrir le remodelage délétère du ventricule.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :

Articles sur le même thème
publicité
publicité