

Publié le 15 nov 2017Lecture 11 min
Mise au point sur l’amylose cardiaque
Diane BODEZ, Thibault DAMY, centre de référence national amyloses cardiaques ; Filière Cardiogen ; Service de cardiologie, Unité Insuffisance Cardiaque – Amylose, CHU Henri Mondor, Créteil - sos.amylosecoeur@aphp.fr
Journées française de l'insuffisance cardiaque
Les amyloses cardiaques regroupent des maladies aboutissant à l’infiltration du myocarde par des protéines fibrillaires. Cette pathologie était jusqu’alors méconnue et sous-estimée, ce qui induisait une absence de diagnostic ou un retard de la prise en charge. Les premières données de prévalence démontrent que les amyloses cardiaques à transthyrétine (TTR) dans sa forme héréditaire pourraient représenter jusqu’à 5 % des cardiomyopathies hypertrophiques (CMH) et dans sa forme sénile en cause dans 13 % des insuffisances cardiaques à fraction d’éjection (FEVG) préservée) et être associée à 16 % des rétrécissements aortiques.
Le pronostic naturel de ces maladies est sombre, mais, la nécessité d’adapter le traitement cardiologique qui s’oppose à l’insuffisance cardiaque classique, la possibilité de greffe cardiaque, l’efficacité des chimiothérapies pour les amyloses AL et surtout le développement de nouveaux traitements spécifiques pour stopper ou retirer les dépôts justifient d’en faire le diagnostic.
Pour ces raisons, il est aujourd’hui essentiel pour le cardiologue de pouvoir reconnaître les signes évocateurs d’amylose cardiaque et d’en connaître les stratégies diagnostiques et thérapeutiques.
Différents types d’amylose cardiaque
Les amyloses cardiaques sont un groupe hétérogène de maladies ayant comme point commun l’accumulation extracellulaire de dépôts protéiques : les fibrilles amyloïdes. Ces dépôts peuvent être localisés ou systémiques avec atteinte de multiples organes : cœur, rein, système nerveux périphérique, tube digestif, glandes salivaires, graisse sous-cutanée, canaux carpiens, tissus mous de la sphère ORL. L’atteinte cardiaque détermine le pronostic. Parmi la vingtaine de précurseurs amyloïdes identifiés, deux protéines sont responsables de la plupart des atteintes cardiaques : les chaînes légères (CL) d’immunoglobuline impliquées dans les amyloses AL, et la transthyrétine (TTR) impliquée dans les amyloses à TTR, qui peuvent être héréditaires (TTR-h, ou familiales) en cas de mutation de la TTR, ou sauvages (TTR-wt, ou séniles) en l’absence de mutation.
Les amyloses cardiaques sont classées par la Société européenne de cardiologie comme cardiomyopathies restrictives (CMR) et/ou hypertrophiques (CMH) selon la présentation phénotypique, et sont un véritable diagnostic différentiel des CMH sarcomériques.
Amylose à chaînes légères (AL)
Les amyloses AL sont la conséquence d’une anomalie monoclonale B, généralement une gammapathie monoclonale de signification indéterminée (MGUS) ou un myélome (qui peut être indolent). Si la prévalence des gammapathies monoclonales augmente avec l’âge (jusqu’à 10 % après 80 ans), l’incidence de l’amylose AL est d’environ 500 cas/an en France. C’est une maladie systémique dont les atteintes les plus fréquentes sont rénale (environ 50 %), hépatique (16 %), digestive (10 %) et cardiaque (60 %).
Amylose à transthyrétine héréditaire (TTR-h)
Plus de 100 mutations de la TTR ont été identifiées comme pourvoyeuses d’amylose, avec des phénotypes variés allant d’une atteinte cardiaque exclusive à une atteinte neurologique pure, avec divers degrés de formes mixtes. La mutation est identifiée par un test génétique qui exclut le diagnostic d’amylose TTR-h s’il est normal. Les mutations fréquemment retrouvées en France en cas d’atteinte cardiaque sont : V122I (forme cardiaque), S77T (forme mixte), L111M et T60A. La mutation V122I la plus fréquente : plus de 3 % des Afro-Américains en sont porteurs, avec une pénétrance incomplète (existence de porteurs sains). La mutation V30M est associée à une forme neurologique (début précoce) ou mixte (forme tardive). La transmission se fait sur un mode autosomique dominant, avec un âge de début qui peut être très tardif dans les formes cardiaques, généralement après 60 ans, et jusqu’à plus de 90 ans.
Face à une amylose à TTR, le test génétique est indispensable quel que soit l’âge du patient, seul moyen de différencier une amylose TTR-wt d’une forme héréditaire, aux lourdes conséquences familiales.
Amylose à transthyrétine sauvage (TTR-wt)
L’amylose TTR-wt, ou sénile, est majoritairement diagnostiquée chez des hommes (> 90 %), possiblement en raison d’un âge de début plus tardif et d’un dépistage moindre chez les femmes. Sa prévalence augmente avec l’âge, (jusqu’à 25 % de dépôts amyloïdes cardiaques après 85 ans dans certaines séries autopsiques).
Le typage de l’amylose est fondamental et parfois complexe, notamment chez le sujet âgé puisque statistiquement 10 % des amyloses TTR-wt sont associées à une gammapathie qui n’est pas responsable de l’amylose.
Signes évocateurs à reconnaître
De façon générale, une amylose cardiaque doit toujours être évoquée en cas d’insuffisance cardiaque associée à une hypertrophie myocardique ou insuffisance cardiaque à FEVG préservée.
Manifestations cardiaques
Cliniquement, l’amylose cardiaque se manifeste le plus souvent par un tableau d’insuffisance cardiaque avec prépondérance des signes d’insuffisance cardiaque droite. Des lipothymies ou syncopes peuvent être liées à des troubles du rythme, de la conduction, ou à une hypotension orthostatique (dysautonomie vasculaire).
Une dyspnée d’effort sans surcharge devra faire rechercher une insuffisance chronotrope (absence d’accélération de la fréquence cardiaque à l’effort, puis baisse de la fréquence cardiaque de repos). Cette « bradycardie relative » aggrave la diminution du débit cardiaque déjà due à la diminution du volume d’éjection systolique. De plus, l’amylose induit un état d’hypercoagulabilité et une hypocontractilité atriale responsables d’une augmentation du risque thromboembolique veineux et artériel, même sans trouble du rythme, qui justifie de discuter systématiquement une anticoagulation efficace, et de rechercher systématiquement un thrombus intracardiaque avant une cardioversion.
L’ECG est anormal dans 90 % des cas. Le classique microvoltage (amplitude des QRS < 5 mm dans les dérivations périphériques, < 10 mm dans les précordiales) ou un voltage normal discordant d’avec l’hypertrophie échographique, est très évocateur mais inconstant et plus souvent observé en cas d’amylose AL que TTR.
L’aspect de pseudo-nécrose dans le territoire antéro-septal est aussi fréquent (50 %), et peut conduire à tort au diagnostic de cardiopathie ischémique, renforcé par une élévation fréquente de la troponine. Les troubles conductifs, BAV1 et bloc de branche, sont observés dans plus d’un cas sur cinq, particulièrement dans les amyloses TTR-wt, et doivent faire discuter l’implantation d’un pacemaker. Les biomarqueurs cardiaques troponine et/ou peptides natriurétiques sont élevés, à des niveaux pouvant contraster avec les symptômes et les anomalies imageries modérés.
L’échographie cardiaque (figure 1) montre un épaississement des parois myocardiques (« hypertrophie »), qui peut être modéré (12-15 mm) en cas d’amylose AL, et parfois majeur avec authentique obstruction intra-myocardique dans certaines amyloses TTR. Rarement, l’épaisseur myocardique peut être normale. Le classique aspect granité et brillant du myocarde n’est plus spécifique avec les récents échographes et l’utilisation de la double harmonique. L’épaississement des valves mitrales et tricuspides est fréquent dans les amyloses TTR ; celui du septum inter-atrial est évocateur mais plus rare. Un épanchement péricardique est plus souvent retrouvé dans l’amylose AL. La FEVG est préservée jusqu’à un stade tardif, mais la fonction systolique étudiée en Doppler tissulaire ou analyse de déformation est précocement altérée, d’abord sur les segments basaux (gradient base-apex). Toutefois, un tiers des amyloses TTR-wt ont une FEVG < 35 %.
Figure 1. Amylose cardiaque en échographie. A. Vue parasternale grand axe avec hypertrophie biventriculaire, lame d’épanchement péricardique. B. Altération du strain global longitdinal en cocarde (gradient base-apex).
La triade hypertrophie biventriculaire, profil restrictif et épanchement péricardique est très évocatrice d’amylose cardiaque, mais signe un stade avancé. Pour changer le pronostic, il est impératif de faire le diagnostic plus précocement. L’altération du strain global avec un gradient base-apex est très spécifique, notamment quand la FEVG est préservée.
Manifestations extracardiaques
Certaines manifestations extracardiaques doivent faire suspecter l’amylose :
– TTR-wt : canaux carpiens précédant l’atteinte cardiaque de 5 à 10 ans, canal lombaire étroit, surdité de perception ;
– TTR-h : neuropathie périphérique, dysautonomie vasculaire ;
– AL : atteinte des tissus mous (macroglossie, ecchymoses périorbitaires, dysphonie, fragilité cutanée, pseudo-hypertrophie musculaire), atteinte rénale (glomérulaire) et plus rarement hépatique.
L’atteinte dysautonomique peut être observée dans tous les types d’amylose (moins fréquente dans les TTR-wt) : hypotension orthostatique, syncope, insuffisance chronotrope, de troubles digestifs, incontinence, dysfonction érectile, agueusie.
Principes de la stratégie diagnostique
(figure 3)
Figure 3. Stratégie diagnostique devant une suspicion d’amylose cardiaque.
En cas de suspicion d’amylose cardiaque, la stratégie diagnostique visera à confirmer le diagnostic positif d’amylose et à déterminer le type d’amylose. Le diagnostic positif de certitude repose sur une preuve histologique qui doit être recherchée systématiquement : biopsie des glandes salivaires accessoires, canal carpien, biopsie sous-cutanée, biopsie rectale, ou orientée par une atteinte d’organe (rénale, hépatique, gastrique, cardiaque, etc.). L’anatomopathologie pourra mettre en évidence les dépôts d’amylose (rouge Congo positifs, avec dichroïsme et biréfringence jaune/vert en lumière polarisée), puis les typer par immunohistochimie ou immunofluorescence (nécessité d’un prélèvement congelé). L’histologie pourra différencier une amylose AL d’une TTR, mais pas une TTR-h d’une TTR-wt.
La preuve histologique étant parfois difficile à obtenir malgré des prélèvements répétés, le diagnostic sera alors posé sur un faisceau d’arguments :
IRM cardiaque : confirmera certains éléments échographiques (FEVG, épanchement péricardique, épaississement pariétal diffus), recherchera un thrombus intracardiaque, et pourra renforcer la suspicion diagnostique d’amylose après injection de gadolinium : wash-out plus rapide rendant difficile le réglage du temps d’inversion, et rehaussement tardif (> 95 %) typiquement diffus pouvant toucher toutes les structures cardiaques. L’augmentation des valeurs de T1 mapping est décrite en cas d’amylose cardiaque mais peu utilisée en routine. Rappelons que l’IRM peut être mise en défaut, et qu’elle ne permet en aucun cas le typage de l’amylose.
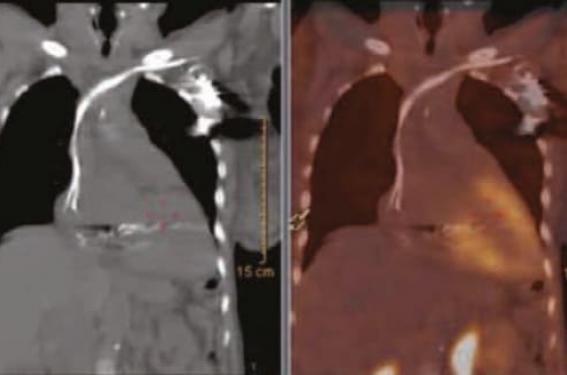
Scintigraphie osseuse (99mTc-DPD,99mTc-HMPD, 99mTc-MPD, 99mTc-PYP) (figure 2), d’accès facile, primordiale en raison d’une fixation myocardique en cas d’amylose à TTR, mais pas AL. Une scintigraphie positive confirmera donc l’amylose, et aussi le type TTR (sans distinguer TTR-h et TTR-wt). Une scintigraphie négative n’éliminera pas une amylose AL (fixation faible ou absente). Ainsi, si la scintigraphie osseuse est fortement positive (grade 2-3), et que le bilan immuno-hématologique plasmatique et urinaire ne retrouve pas de gammapathie, le diagnostic d’amylose TTR peut être posé sans biopsie.
Figure 2. Amylose cardiaque TTR en scintigraphie osseuse. A. Corps entier avec traceur HMDP de face et de dos. B. Scintigraphie couplée au scanner montrant précisément la localisation myocardique du traceur.
Dans tous les cas, il convient d’éliminer une amylose AL pour laquelle un traitement spécifique existe et doit être instauré en urgence. Chez le sujet âgé avec gammapathie clonale (fréquente, 10 %), une preuve histologique est nécessaire pour différencier AL de TTR-wt. En cas de forte suspicion d’amylose AL, et de biopsie extracardiaque (glande salivaire) négative, une biopsie ciblée sur les organes atteints (myocarde) sera rapidement envisagée afin d’éviter la multiplicité des biopsies périphériques de sensibilité moyenne qui retarderont de plusieurs semaines la mise en route du traitement.
Les grands principes thérapeutiques, généraux et spécifiques
Le typage de l’amylose est indispensable pour instaurer un traitement spécifique. Le typage histologique est indispensable avant toute chimiothérapie pour amylose AL.
Principes généraux
En raison du trouble de compliance myocardique, et d’une insuffisance chronotrope fréquente qui aggrave la baisse du débit cardiaque, les traitements bradycardisants sont à proscrire, particulièrement les bêtabloquants. La digoxine est aussi à éviter en raison d’une potentielle accumulation dans les dépôts d’amylose qui pourrait majorer les troubles conductifs. L’amiodarone est donc le seul traitement antiarythmique non contre-indiqué, et peut être utilisé à visée de contrôle du rythme ou de la fréquence.
Les autres traitements classiques de l’insuffisance cardiaque, IEC ou ARA2, sont à éviter en raison du risque d’hypotension, mais peuvent être maintenus à visée néphroprotectrice lorsqu’ils sont bien tolérés.
Les diurétiques sont donnés pour contrôler les signes congestifs (diurétiques de l’anse ± épargneurs de potassium), mais à manier avec précaution en raison du risque d’hypotension orthostatique.
Une anticoagulation efficace doit être instaurée en cas d’antécédent thromboembolique, de profil mitral restrictif, de dysfonction auriculaire sévère ou de contraste spontané échographique. Enfin les dysautonomies invalidantes peuvent être traitées par bas de contention, midodrine, et mesures hygiéno-diététiques importantes.
L’implantation d’un pacemaker devra être systématiquement discutée à partir des données ECG et d’un Holter des 24 heures. Les indications sont habituellement élargies (BAV du premier degré, bloc de branche, syncopes inexpliquées, HV > 70 ms) mais ne reposent encore sur aucune recommandation scientifique. L’implantation d’un défibrillateur en prévention primaire peut être décidée sur l’existence d’une dysfonction systolique établie en strain (FEVG prise en défaut), mais là encore le niveau de preuve reste faible.
En cas d’insuffisance cardiaque terminale, l’assistance circulatoire ventriculaire gauche de longue durée (LVAD) n’est pas indiquée (petite taille des ventricules, fréquente dysfonction biventriculaire, sur-risque thrombotique, potentielles complications hémorragiques par atteinte vasculaire amyloïde). Par contre, la transplantation cardiaque, parfois précédée d’ECMO, peut être une option chez des patients jeunes avec atteinte cardiaque isolée.
Traitements spécifiques
Les traitements actuels de l’amylose AL visent à réduire la production des chaînes légères par chimiothérapies. Le pronostic étant directement déterminé par l’atteinte cardiaque, le type de traitement proposé dépend du stade de gravité initial (classification de la Mayo Clinic basée sur l’élévation des biomarqueurs cardiaques). La réponse hématologique est évaluée sur l’évolution du taux de chaînes légères libres. En cas d’amylose cardiaque, l’élévation des biomarqueurs est fréquente et le traitement de référence est aujourd’hui bortézomib – cyclophosphamide – endoxan (VCD). La greffe de cellules souches privilégiées par certaines équipes est rarement possible en cas d’atteinte cardiaque (surmortalité importante).
Concernant l’amylose TTR-h, des traitements limitant les dépôts de TTR par stabilisation de la protéine sous forme non fibrillaire (tafamidis) sont disponibles pour les indications neurologiques, mais toujours à l’étude en ce qui concerne l’atteinte cardiaque (essais de phase 3). La greffe hépatique, remplaçant la synthèse hépatique de la TTR mutée par une TTR normale, fait partie des possibilités thérapeutiques en cas d’amylose TTR-h, mais est de moins en moins réalisée depuis le développement des traitements médicamenteux. En cas d’indication de transplantation cardiaque, une greffe combinée cœur-foie peut être proposée. Dans tous les cas, une prise en charge familiale en conseil génétique est indispensable pour toutes les amyloses TTR-h.
De nombreux traitements prometteurs sont à l’étude : antiplasmocytes sécréteurs de chaînes légères (amylose AL), stabilisateurs de protéines à l’état fibrillaire, molécules empêchant la production des précurseurs protéiques TTR, ou ciblant directement les dépôts d’amylose (anticorps antifibrilles). L’histoire de l’amylose cardiaque ne fait que commencer et le pronostic de cette maladie sera sans nul doute prochainement transformé.
En pratique
Suspecter une amylose cardiaque devant toute cardiopathie hypertrophique et/ou insuffisance cardiaque à FEVG préservée.
Savoir reconnaître les signes évocateurs d’amylose cardiaque : discordance hypertrophie échographique/microvoltage ECG, discordance FEVG préservée/élévation majeure des bio marqueurs cardiaques, discordance FEVG préservée/altération du strain global avec gradient base apex, signes extracardiaques (canal carpien, neuropathie périphérique, ecchymoses périorbitaires et macroglossie), histoire familiale.
Urgence diagnostique pour adaptation du traitement : scintigraphie aux traceurs osseux pour les amyloses à TTR, preuve histologique pour l’amylose AL.
Prise en charge en dehors des recommandations classiques de l’insuffisance cardiaque : contre-indication aux bêtabloquants et bradycardisants, anticoagulation efficace à discuter systématiquement, prévention des fréquentes complications rythmiques et conductives.
Pronostic modifié par les nouvelles thérapeutiques et la possibilité de greffe cardiaque pour des patients sélectionnés.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :

Articles sur le même thème
publicité
publicité