


Publié le 01 sep 2009Lecture 13 min
Antiagrégants plaquettaires et angioplastie coronaire - Session du GACI organisée dans le cadre du Printemps de la cardiologie
E. MILLARA, Suresnes
Printemps de la cardiologie
Thrombose et inflammation de plaques
D’après M. Sirol (Paris)
La plaque d’athérome évolue naturellement vers l’érosion ou la rupture, provoquant des thromboses partielles ou complètes. Le délai entre la survenue d’une rupture de plaque et l’apparition d’événements cliniques s’étend de quelques minutes à plusieurs mois et dans 50 % des cas, lors de la revascularisation, les thrombus sont constitués depuis plus de 3 jours et même dans 16 % des cas, depuis plus de 5 jours, avec une organisation déjà constituée. Or, l’ancienneté du thrombus lors de la revascularisation est un facteur de mortalité important : 16 % à 4 ans contre 7 % pour un thrombus frais. En outre, dans 25 % des cas de rupture symptomatique, il existe au moins deux plaques rompues.
Les facteurs clés de rupture sont la vulnérabilité des plaques, avec un cœur lipidique important et une chape fibreuse fine, la présence d’une thrombose partielle et l’existence d’une inflammation de la plaque et/ou une néovascularisation.
Inflammation et thrombose entretiennent des rapports étroits et réciproques. Les biomarqueurs de l’inflammation (IL6, TNF-·, MCP-1, P-sélecine, etc.) s’élèvent des années avant un infarctus myocardique, et certains sont hautement prédictifs de récidive d’infarctus.
En pathologie cardiovasculaire, le rôle de l’inflammation a, en particulier, été mis en évidence par les études PROVE-IT et TIMI-22, rapportant que la CRP ultrasensible représente un facteur prédictif majeur d’événements coronariens et de mortalité, plus puissant que le LDL-cholestérol.
En effet, au niveau de la plaque athéromateuse, la dysfonction endothéliale permet l’internalisation des monocytes, contribuant au développement du volume de la plaque. Histologiquement, il apparaît que les plaques rompues sont très riches en cellules inflammatoires, principalement macrophages mais aussi lymphocytes T, et pauvres en cellules musculaires lisses. L’évolution des monocytes en cellules spumeuses productrices de cytokines pro-inflammatoires génère une inflammation au sein de la plaque, et on observe que les marqueurs de l’inflammation sont plus élevés dans l’angor instable que dans l’angor stable.
Les cytokines pro-inflammatoires exercent un ensemble d’actions très délétères conduisant à la thrombose : elles renforcent la dysrégulation de l’endothélium et inhibent ses propriétés antiagrégantes ; elles augmentent le recrutement des monocytes et contribuent ainsi au développement de la lésion et à l’augmentation de son inflammation ; en outre, ces cytokines stimulent les métalloprotéinases qui dégradent la matrice extracellulaire, réalisant un grignotage progressif de la chape fibreuse qui rend la plaque vulnérable et favorise sa rupture ; enfin, elles stimulent l’apoptose, notamment à l’échelon des cellules musculaires lisses, ce qui contribue à la fragilisation de la chape.
L’inflammation résulte d’un déséquilibre entre médiateurs pro-inflammatoires et anti-inflammatoires. Ainsi, il a été montré expérimentalement que des souris déficientes en récepteur de l’interféron gamma développaient une plaque plus riche en collagène et plus stable que des souris non déficientes. L’expression de l’interleukine 18 est plus importante chez les coronariens symptomatiques, par comparaison aux coronariens asymptomatiques, et les taux circulants d’IL18 sont associés à un pronostic d’autant moins favorable qu’ils sont élevés.
Inversement, l’interleukine 10 est une cytokine anti-inflammatoire : expérimentalement, des souris déficientes en récepteur de l’IL10 développent des plaques d’athérome plus larges et plus lipidiques que des souris non déficientes, et les taux circulants d’IL10 sont moindres chez les patients présentant un angor instable, par comparaison avec des patients stables.
Les patients souffrant d’un infarctus récent ou d’angor instable ont également un taux plus élevé de microparticules circulantes que les patients atteints d’angor stable. Ces microparticules de bicouche lipidique sont émises par les cellules inflammatoires apoptotiques et sont hautement athérogènes, activant l’expression de molécules d’adhésion au niveau de l’endothélium. Un grand nombre de ces microparticules sont libérées lors de la rupture de plaque, générant une accélération du processus thrombogène (figure 1).
Figure 1. Athérosclérose et inflammation.
Le rôle des plaquettes dans la formation du thrombus est bien connu. Les plaquettes activées déclenchent la formation du caillot et le densifient. Lorsque le thrombus est partiel et non occlusif, la cicatrisation de la plaque rompue intègre le thrombus et contribue également à la progression de la plaque athéromateuse. Le facteur tissulaire, exprimé notamment par les macrophages apoptotiques par l’intermédiaire de l’activité des caspases, favorise la formation de thrombus.
En pratique
La prévention de la thrombose chez le patient athéromateux passe nécessairement par le contrôle de l’inflammation au niveau de la plaque, et c’est sans doute par un tel mécanisme que passe l’effet préventif des statines.
Antiagrégants plaquettaires et risque thrombotique au long cours
D’après T. Cuisset (Marseille)
Après une angioplastie coronaire, l’administration d’aspirine à vie constitue une recommandation de classe 1. Il semble qu’une dose supérieure à 100 mg/j ne soit associée à aucun bénéfice en termes de réduction du risque ischémique, mais comporte en revanche un risque hémorragique dose dépendant. Dans l’attente des nouvelles données sur cette question, qui seront apportées par l’étude CURRENT-OASIS 7 en association au clopidogrel, les recommandations actuelles préconisent une dose d’entretien comprise entre 75 et 100 mg.
Les véritables résistances à l’aspirine sont peu fréquentes. La notion de résistance à l’aspirine est surestimée par 2 facteurs : la grande variabilité de tests plaquettaires utilisés, plus ou moins spécifique de l’effet de l’aspirine mais aussi une mauvaise observance du patient, qui constitue la cause principale de cette « résistance », comme le montre sa fréquence plus élevée chez les patients ambulatoires que chez les patients hospitalisés.
Le recours à la bithérapie antiplaquettaire s’est développé pour réduire l’incidence élevée des thromboses de stent et les événements récurrents après syndrome coronarien aigu. La grande question actuelle reste la durée optimale d’administration de la thiénopyridine : une durée de 12 mois est recommandée après tout syndrome coronaire aigu. Après implantation d’un stent actif, les recommandations divergent selon leur origine : au moins 12 mois pour les sociétés savantes américaines (ACC/AHA/SCAI 2007) contre 6 à 12 mois pour l’ESC. En effet, il apparaît que l’arrêt prématuré de la thiénopyridine accroît la mortalité à 1 an, avec une relation particulièrement marquée lorsque cet arrêt intervient dans les 6 premiers mois. Toutefois, après 6 mois, l’incidence des thromboses de stent ne semble pas significativement différente selon que le patient poursuive ou non le traitement par thiénopyridine. Ces données issues de deux registres distincts et concordants suggèrent ainsi qu’une bithérapie de 6 mois serait suffisante, tandis que les recommandations préconisent actuellement 1 an. Plusieurs études randomisées sont en cours dans le but de définir précisément la durée optimale de cette bithérapie, notamment les études françaises ARCTIC, comparant des durées de bithérapie de 12 et 18 mois et ITALIC, comptabilisant la mortalité et la morbidité ischémique sur 1, 2 et 3 ans selon que la thiénopyridine est arrêtée après 6 mois ou poursuivie. Une étude similaire est en cours en France sous l’égide de la SFC, l’étude OPTIDUAL pilotée par Gérard Helft. L’étude internationale ISAR-SAFE comparera pour sa part une durée de traitement de 6 et 12 mois, avec comptabilisation des événements majeurs 15 mois après l’implantation du stent.
Dans l’année suivant une thrombose de stent confirmée par angiographie, la mortalité cardiovasculaire est de 10 % et la récidive de la thrombose de 15 %. Mais l’étude ZEST a montré que les thromboses de stent ne représentent que 10 % des événements ischémiques survenus. Le risque de thrombose n’est pas seulement lié au stent, mais également au niveau de risque du patient : selon les données de l’étude CHARISMA, la bithérapie par aspirine + clopidogrel n’apporte pas de bénéfice en termes d’événements athérothrombotiques par comparaison à l’aspirine seule si l’on considère la population totale. Par contre, le bénéfice est significatif au sein des sous-populations à haut risque en prévention secondaire (antécédents d’infarctus, d’AVC ou d’artériopathie périphérique symptomatique). La durée de la bithérapie « idéale » n’est probablement pas la même d’un patient à l’autre et il conviendra de l’adapter aux risques ischémique et hémorragique du patient.
Pour le risque hémorragique, il concerne aussi plus particulièrement une population fragile, âgée de plus de 75 ans, présentant des antécédents de saignements sous antiagrégants, d’AVC ou une insuffisance rénale, il est probablement judicieux d’adapter la durée de la bithérapie antiagrégante au profil du patient : au delà de 12 mois en cas de profil à haut risque thrombotique (pluritronculaires, récidives de SCA, diabétiques, post-STEMI…) et plus courte en cas de profil à haut risque de saignement.
L’arrivée de nouveaux antiplaquettaires va très prochainement encore bouleverser la donne.
L’étude TRITON-TIMI 38 réalisée chez 13 600 patients ayant bénéficié d’une angioplastie sur syndrome coronarien aigu a montré après 15 mois de suivi une incidence significativement moindre de décès, infarctus et AVC chez les patients traités par prasugrel (dose de charge de 60 mg puis dose d’entretien de 10 mg), comparativement au clopidogrel (dose de charge de 300 mg puis dose d’entretien de 75 mg), avec une incidence du critère composite de 9,9 % versus 12,1 %. Cette efficacité clinique supérieure s’accompagne cependant d’une augmentation des saignements (2,4 % versus 1,8 %), une augmentation comparable à celle qui avait été observée sous clopidogrel avec l’étude CURE par rapport au placebo.
TRITON a permis d’identifier les patients présentant un sur-risque de saignement (patients ≥ 75 ans, < 60 kg ou avec un antécédant d’AVC) et à l’inverse les patients à haut risque avec un bénéfice clinique net très favorable au prasugrel (diabétiques, STEMI).
En pratique
La réponse au clopidogrel présente une certaine variabilité, avec un taux de récidives ischémiques et de thromboses de stent significativement plus élevé chez les non-répondeurs. Une nouvelle thiénopyridine, représentée par le prasugrel, bientôt disponible dans la prise en charge des SCA avec angioplastie, a démontré un bénéfice anti-thrombotique supplémentaire comparé au clopidogrel, mais aussi un surrisque hémorragique dans certaines populations à prendre en considération. Particulièrement intéressant chez les patients à haut risque thrombotique avec un bénéfice démontré sur 1 an, le prasugrel à la dose étudiée dans TRITON devra être évité chez les patients à risque hémorragique : plus de 75 ans, moins de 60 kg ou présentant des antécédents d’AVC.
La réponse aux nouvelles thiénopyridines
D’après G. Cayla (Nîmes)
La ticlopidine a représenté la 1re génération des thiénopyridines utilisée dans les années 1990 après angioplastie. Le clopidogrel l’a remplacé à partir des années 2000 et la 3e génération de thiénopyridines, représentée par le prasugrel, devrait être disponible d’ici 2010.
Les thiénopyridines sont des pro-drogues, nécessitant une transformation en métabolites actifs dans l’organisme. Ainsi, l’existence de patients non répondeurs au clopidogrel, présentant un risque accru d’évènements cardiovasculaires, s’explique-t-elle en partie par un polymorphisme génétique de ses voies métaboliques, mais également par des facteurs cliniques, tels que l’existence d’un diabète et par des facteurs cellulaires.
La pharmacologie des thiénopyridines passe par les phénomènes classiques d’absorption et de distribution, puis subit un métabolisme selon deux phases : la phase I d’hydrolyse par des estérases et d’oxydation se déroule au niveau des cytochromes, et produit les métabolites actifs. La phase II de conjugaison permet ensuite leur élimination.
Le clopidogrel est hydrolysé à 85 % par les estérases intestinales, et seule une fraction de 15 % est absorbée puis transformée en métabolite actif au niveau du cytochrome P450 hépatique, par plusieurs cytochromes oxydases, la CYP 2C19 étant principalement impliquée dans les deux étapes de ce métabolisme.
Le prasugrel est beaucoup mieux absorbé et subit une seule étape métabolique au niveau du cytochrome P450, mettant également en jeu plusieurs cytochromes oxydases avec une moindre dépendance vis-à-vis de la CYP 2C19.
La différence pharmacologique entre ces deux molécules se situe principalement au niveau de la quantité de métabolites actifs disponibles (réponse pharmacocinétique). En effet, en termes de pharmacodynamie, l’effet d’inhibition plaquettaire respectif des métabolites actifs est identique, qualitativement et quantitativement. Il a été mis en évidence une génération de métabolites actifs plus importante et plus rapide sous prasugrel, comparativement au clopidogrel, permettant un effet biologique d’inhibition plaquettaire également plus puissant et plus rapide, cette différence étant maintenue dans le temps en phase d’entretien, y compris en comparaison aux doses élevées de clopidogrel. (figure 2)
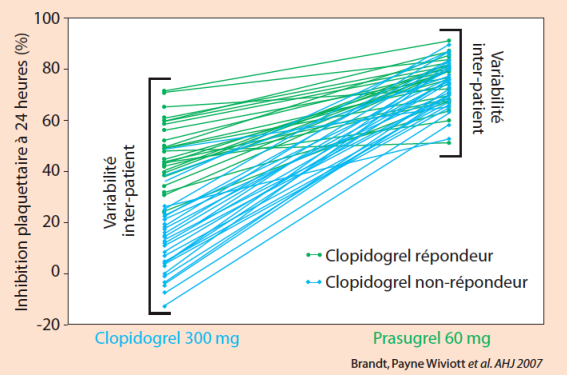
Elle se traduit également par une moindre variabilité sous prasugrel par rapport au clopidogrel. (figure 3)
Figure 2. PRINCIPLE TIMI-44 : taux d’inhibition plaquettaire.
Figure 3. Variabilité inter-individuelle sous prasugrel vs clopidogrel. Témoins sains = 68.
Le niveau d’activité du cytochrome 2C19 peut être l’objet d’interactions médicamenteuses et notamment d’une inhibition par certains médicaments tels que les IPP, et il fait l’objet d’un important polymorphisme génétique. Il joue un rôle déterminant dans le métabolisme du clopidogrel, mais beaucoup plus modeste dans celui du prasugrel. Il a ainsi été observé une réduction de 32 % de la réponse pharmacocinétique au clopidogrel et de 9 % de la réponse pharmacodynamique chez des sujets porteurs d’une mutation homozygote du CYP 2C19, tandis que cette réduction n’est que de 6 % et 1,3 % respectivement pour les réponses au prasugrel. En effet, la variabilité de réponse au prasugrel s’avère beaucoup plus faible qu’avec le clopidogrel, avec un taux de répondeurs supérieur indiquant une moindre sensibilité aux variations génétiques du CYP 2C19.
Il existe une bonne corrélation entre la présence de la mutation et la survenue d’événements cliniques d’origine thrombotique sous clopidogrel, tandis qu’une telle corrélation n’est pas mise en évidence avec le prasugrel. (figure 4)
Figure 4. Effets mutation 2C19*2 comparaison Clopidogrel/Prasugrel.
En pratique
Le prasugrel apparaît comme beaucoup moins dépendant du polymorphisme génétique du CYP 2C19 que le clopidogrel, et supérieur en termes d’intensité et de rapidité de l’inhibition plaquettaire, avec un meilleur taux de patients répondeurs. Le mécanisme d’action des deux produits est identique, consistant en une inhibition plaquettaire irréversible, et leur durée d’action est identique, correspondant à la durée de vie des plaquettes.
Les tests d'agrégation plaquettaire en angioplastie : quel avenir ?
D’après P. Barragan (Ollioules)
Le risque de thrombose après implantation d’un stent est maximal dans les 30 premiers jours, nettement moindre après 6 mois et à 1 an, mais toujours grave. L’étude TRITON a montré la supériorité du prasugrel sur le clopidogrel en termes d’événements ischémiques, avec un bénéfice maximal du prasugrel au cours des 30 premiers jours, qui persiste sur toute la durée de l’étude. Malgré tout, il persiste un taux apparemment incompressible de 5 % d’accidents ischémiques dans les 3 premiers jours.
Il est important pour le clinicien de pouvoir suivre le niveau d’inhibition plaquettaire de façon simple et rapide.
Sur le plan phénotypique, trois tests permettent à l’heure actuelle d’apprécier l’inhibition plaquettaire : le VASP, qui représente le « gold standard » pour évaluer la réponse pharmacologique aux thiénopyridines, l’agrégométrie optique et le VERIFYNOW qui présentent des limites dans le contexte d’angioplastie.
Sur le plan génétique, il est démontré que les mutations portant sur le CYP 2C19 réduisent la réponse au clopidogrel, avec un doublement du risque d’événements péjoratifs cumulés 1 an après l’angioplastie chez les sujets présentant une mutation homozygote. Cependant, la réalisation de tests génétiques présente un intérêt limité en pratique, sachant que les variants mutés constituent 30 % à 55 % de la population, soit un groupe à risque beaucoup trop important pour pouvoir pratiquer un dépistage génétique à un coût acceptable.
Face à ces difficultés de monitoring en routine, le prasugrel présente un intérêt car moins sensible que le clopidogrel aux mutations du CYP2C19 ; se posera la question du monitoring pour clopidogrel, notamment chez les patients qui ne tirent pas un réel bénéfice du prasugrel, à savoir les patients de plus de 75 ans, de moins de 60 kg ou présentant un antécédent d’AVC ou d’augmenter les doses de clopidogrel à 900 mg (dose de charge)/150 mg (dose d’entretien).
Plusieurs études indiquent par ailleurs qu’il existe également des résistances à l’aspirine - entre 5 et 25% des patients environ - et que de telles résistances augmentent très significativement le risque cardiovasculaire, sans bénéfice de l’adjonction de clopidogrel chez ces patients. Il n’y a pas de test rapide pour dépister les résistances à l’aspirine, et les résultats sont fortement dépendants du test utilisé (thromboxane sérique ou urinaire, agrégométrie, PFA-100 ou Verify Now).
Il existe actuellement un besoin non couvert de tests simples et rapides permettant d’apprécier le niveau d’inhibition plaquettaire chez chaque patient, afin de pouvoir optimiser le traitement anti-agrégant de façon personnalisée, avec pour objectif un risque minimal de thrombose de stent, d’événements ischémiques et d’accidents hémorragiques.
En pratique
Le traitement antiagrégant après angioplastie doit être personnalisé selon le profil du patient qu’il faut impérativement déterminer en fonction du risque thrombotique ou à haut risque hémorragique. La durée de la bithérapie antiagrégante ainsi que le choix de la thiénopyridine doivent être adaptés en fonction de ce profil. L’arrivée prochaine de la 3e génération de thiénopyridines, comportant une efficacité renforcée mais également un surrisque hémorragique, accroît le besoin de surveillance du niveau d’inhibition obtenu individuellement chez chaque patient, et le développement de tests simples, fiables et rapides constitue un des enjeux de l’avenir.
D'après une session parrainée par les laboratoires Daiichi-Sankyo et Lilly
Sous la présidence de M. Gilard (Brest) et G. Helft (Paris)
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :

Articles sur le même thème
publicité
publicité