



Publié le 18 oct 2011Lecture 10 min
Syncopes à l’exercice chez l’enfant : que faire ?
J.-M. LUPOGLAZOFF1,2, I. DENJOY2,3 1 Hôpital Robert-Debré, Paris 2 Groupe Hospitalier Pitié-Salpêtrière, Paris 3 Hôpital Lariboisière, Paris
La perte de connaissance dans un contexte adrénergique d’effort chez l’enfant est une cause fréquente de consultation et de bilan en cardiologie pédiatrique. La syncope d’effort est toujours un événement grave pour lequel il faut mener un bilan le plus complet possible pour poser un diagnostic étiologique et proposer une prise en charge thérapeutique adaptée.
Enquête anamnestique
Dans cette enquête, l’interrogatoire de l’enfant, de la famille et des témoins de la perte de connaissance est un élément important du diagnostic. Il faut déterminer le type d’effort qui a entraîné la perte de connaissance, le déroulement exact des événements : s’agissait-il d’une syncope à l’effort, à la fin de l’effort, au décours de l’effort ou à distance de l’effort ? Dans quel contexte est survenue cette syncope ? L’enfant était-il malade dans les jours précédents ? Avait-il de la fièvre ? S’agissait-il d’une syncope inaugurale. Il faut éliminer, par l’interrogatoire et l’analyse de documents, les causes neurologiques, métaboliques, d’autres anomalies : cardiaques, syndromiques (comme le syndrome de Marfan par exemple).
En plus de détailler les circonstances de la syncope, il faut essayer d’estimer sa durée ; y a-t-il eu des convulsions au décours, des pertes d’urine, des mouvements anormaux, des morsures de langue, des signes cliniques signant une perte de connaissance qui a duré quelques minutes ?
Le même type d’interrogatoire doit s’adresser aux parents et rechercher des antécédents familiaux du même type, ou d’antécédents de syncope ou de mort subite.
Un syndrome de Wolff-Parkinson-White
Pour rechercher ces causes de syncope d’effort chez l’enfant (encadré 1), on doit faire un bilan complet comportant une échocardiographie, un ECG 12 dérivations, un Holter-ECG et une épreuve d’effort si l’enfant a l’âge et la taille suffisants (9-10 ans et 1,35-1,40 m). Chez les enfants plus jeunes, à partir de 5 ans, l’effort peut être évalué lors de l’enregistrement du Holter-ECG. L’ECG de surface et le Holter permettent de rechercher un syndrome de Wolff-Parkinson-White qui peut être une cause de syncope d’effort (encadré 2).
La fibrillation auriculaire sur Wolff-Parkinson-White est exceptionnelle chez l’enfant puisque la fibrillation auriculaire n’existe quasiment pas en cardiopédiatrie.
Lorsqu’une syncope d’effort fait découvrir un syndrome de Wolff-Parkinson-White avec une tachycardie supraventriculaire, il faut interdire le sport, sauf si l’on a une absence de voie accessoire patente, que le traitement bêtabloquant est efficace et que l’épreuve d’effort est négative sous traitement. Dans le cas contraire, c’est-à-dire l’existence d’une voie accessoire patente, la volonté de faire du sport et un âge suffisant au-dessus de 10-12 ans, on peut discuter une ablation par radio-fréquence.
Une cardiomyopathie hypertrophique
L’autre étiologie des syncopes d’effort est la cardiomyopathie hypertrophique dont le diagnostic est fait sur l’ECG et surtout sur l’échocardiographie avec ensuite une analyse génétique, un conseil génétique et une enquête familiale. Ceci débouche sur l’interdiction formelle de compétition, interdiction de sport, sauf chez les enfants asymptomatiques ayant des formes familiales modérées (absence de mort subite familiale, mutation génétique bénigne). Le suivi sera annuel avec un ECG, une épreuve d’effort et un Holter ainsi que l’échocardiographie et ECG des parents.
Les canalopathies
À côté de ces deux étiologies, il faut penser aux canalopathies devant une mort subite d’effort avec autopsie négative (2 à 5 % des cas), une syncope d’effort ou durant la plongée ou devant une émotion ou favorisée par une fièvre ou des antécédents familiaux de syncope ou de mort subite d’effort. Les canalopathies induites par l’exercice sont surtout le syndrome du QT long congénital (SQTL) et les tachycardies ventriculaires polymorphes catécholergiques (TVC). Deux autres canalopathies sont moins concernées par ce contexte, le syndrome de Brugada et le syndrome du QT court.
Le syndrome du QT court est une entité relativement récente avec un intervalle QTc ≤ 300 ms, un risque élevé de syncope ou de mort subite par arythmie ventriculaire maligne. Le QTc court est parfois associé à de la fibrillation auriculaire de survenue précoce. Sur le plan génétique, il s’agit de mutations dans les gènes HERG, KCNQ1 ou KCNH2.
Le seul traitement préventif de la mort subite à l’heure actuelle reste l’implantation d’un défibrillateur cardiaque.
Le syndrome de Brugada touche essentiellement l’homme vers l’âge de 40 ans et les formes à révélation pédiatrique sont extrêmement rares, se manifestant le plus souvent par des arrêts cardiaques ou des morts subites dans un contexte fébrile. L’aspect ECG, parfois intermittent, peut être démasqué par un test pharmacologique consistant à l’injection d’un bloqueur des canaux sodiques. L’aspect génétique a été confirmé par l’identification d’une mutation dans le gène SCN5A codant pour un canal sodique cardiaque dans seulement 20 % des cas.
Le SQTL a une prévalence actuelle de 1 sur 4 000 individus. Dans la population française, les formes LQT1 et LQT2 sont les plus fréquentes, respectivement 50 et 45 %. La forme LQT3 est beaucoup plus rare, 5 %, et survient plutôt au repos.
Le diagnostic de QT long est probable si au moins l’un des critères suivants est présent : QTc > 460 ms, QTc > 440 ms et bradycardie ou morphologie anormale de l’onde T ou syncope ou torsade de pointe dans une famille de QT long.
Il existe une dizaine de gènes impliqués dans le syndrome du QT long congénital et le mode de déclenchement des événements rythmiques varie en fonction du défaut génétique. Le plus souvent, la syncope ou l’arrêt cardiaque en cas de LQT1 survient au cours d’un effort, particulièrement dans un contexte émotionnel (natation). Les patients LQT2 présentent des syncopes et des troubles du rythme plutôt lors d’une stimulation auditive ou d’émotion ou lors d’un réveil nocturne.
L’analyse morphologique de l’onde T sur l’ECG montre des aspects morphologiques assez spécifiques des trois principaux gènes connus (figure 1). Dans la forme LQT3, l’intervalle QTc est très allongé avec une onde T tardive et de grande amplitude. Dans la forme LQT2, l’onde T est de faible amplitude et, dans la forme LQT1, qui est la plus fréquente, la morphologie de l’onde T est monophasique avec une base élargie sensiblement normale, ce qui rend son diagnostic plus difficile.
Figure 1. Représentation schématique des 3 formes principales de SQTL avec leurs principales caractéristiques.
L’analyse de l’ensemble des tracés ECG des membres d’une même famille, en particulier les parents chez l’enfant concerné, améliore la sensibilité et la spécificité du diagnostic pour un patient donné.
De même, l’analyse du Holter-ECG est très contributive à la détection de l’anomalie morphologique de l’onde T chez les parents suspects de SQTL. La survenue des symptômes dépend de l’âge, du sexe, de la durée du QTc et du génotype. L’âge du premier événement clinique est plus précoce chez les garçons que chez les filles, mais après 15 ans, les femmes restent plus symptomatiques. D’après Priori et coll., la stratification du risque de premier événement cardiaque avant l’âge de 40 ans, a permis d’établir que les patients à plus haut risque (> 50 %) sont les patients de sexe masculin LQT3, puis les patients LQT1, LQT2 dont la durée du QTc est > 500 ms.
Une fois le diagnostic posé chez l’enfant, la fratrie et les parents du propositus seront vus en consultation multidisciplinaire pour reprendre l’anamnèse de chacun, leur faire un ECG, compléter au moindre doute par un Holter. Le SQTL étant une maladie autosomique dominante, la moitié des apparentés de chaque famille peut être atteinte de SQTL. Lors de la consultation multidisciplinaire, le propositus et les membres de la famille seront prélevés pour une analyse de l’ADN après recueil écrit du consentement éclairé.
Le traitement de référence de la prévention des torsades de pointe est représenté par les bêtabloquants qui doivent être prescrits à tous les patients symptomatiques.
Les données cliniques expérimentales nous ont appris que ce traitement s’adresse avant tout aux patients LQT1, certains patients LQT2 et qu’il est probablement peu adapté aux patients LQT3. Dans les formes qui échappent au traitement bêtabloquant et probablement pour les formes LQT3, le défibrillateur automatique implantable (DAI) trouve sa place. Dans l’avenir, il existera probablement des indications plus gène-spécifiques, en particulier pour ces formes ou dépendant du mode de déclenchement de l’événement cardiaque. La difficulté clinique reste encore la prévention du risque de mort subite pour un individu présymptomatique, génétiquement atteint. Actuellement, dans la mesure où il existe un traitement préventif efficace et bien toléré, les patients génétiquement atteints sont traités préventivement par bêtabloquants.
L’autre grande canalopathie responsable de syncopes d’effort est la tachycardie ventriculaire catécholergique (TVC) qui est caractérisée par des arythmies ventriculaires polymorphes de déclenchement adrénergique. Elles surviennent essentiellement chez des enfants et des adolescents et sont responsables de syncopes et de mort subite, en l’absence de toute anomalie morphologique cardiaque. L’ECG de repos enregistré en dehors des épisodes de tachycardie ventriculaire est souvent normal. La mortalité des TVC en l’absence de traitement est très élevée, atteignant 30 à 50 % avant l’âge de 30 ans. Il existe une corrélation entre l’âge de survenue de la première syncope et la sévérité de l’anomalie avec un pronostic très péjoratif lorsque les pertes de connaissance surviennent très jeunes.
Les bêtabloquants réduisent de façon significative les syncopes et les morts subites, rendant rare l’implantation de défibrillateur implantable.
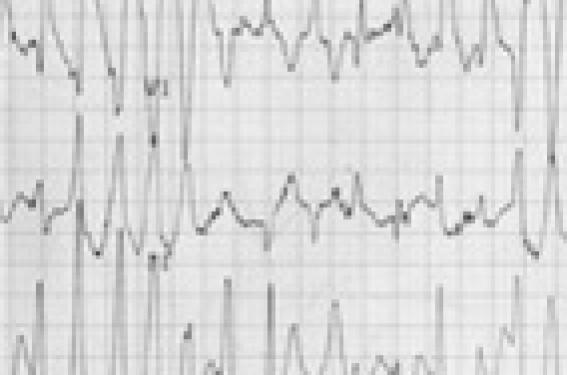
Ce syndrome est caractérisé par des tachycardies polymorphes survenant chez des enfants ou adolescents en l’absence de toute anomalie cardiaque. Elles se produisent dans des circonstances particulières, lors d’efforts violents ou survenant dans un contexte émotionnel et sont responsables de syncopes, voire de morts subites. L’ECG de repos montre dans la moitié des cas une bradycardie sinusale, moins de 60/min, inhabituelle chez l’enfant, un intervalle QT le plus souvent normal. Le Holter, mais surtout l’épreuve d’effort, permet de faire le diagnostic ; le trouble du rythme apparaît comme une séquence caractéristique et stéréotypée au cours de l’effort (figure 2) : tachycardie sinusale, ensuite rythme jonctionnel actif, apparition d’extrasystoles ventriculaires avec une fréquence seuil qui est en général aux alentours de 130-135/min ; tout d’abord monomorphes et isolées, les ESV deviennent polymorphes, le plus souvent à type de retard droit et d’axe variable répétitif ; enfin, aspect de TV bidirectionnelles puis polymorphes ressemblant à des torsades de pointe. Le retour en rythme sinusal se fait à l’arrêt de l’effort, souvent une séquence inverse avec parfois des passages transitoires en tachycardie atriale.
Figure 2. Épreuve d’effort réalisée chez une enfant de 10 ans adressée pour syncope lors de la course. Il existe des salves ventriculaires polymorphes sur fond de rythme sinusal à 150/min.
Les bases génétiques des TVC ont permis de montrer qu’il s’agit d’une mutation transmise de façon autosomique dominante dans le récepteur Ryanodine type 2 (RYR2). Ce récepteur cardiaque a des canaux calciques impliqués dans le relargage du calcium situé dans le réticulum sarcoplasmique des cardiomyocytes. Un autre gène a été identifié CASQ2.
Le traitement de première intention est la mise sous bêtabloquants pour transformer le pronostic de cette maladie. Les posologies utilisées dans la prévention des syncopes d’effort chez les TVC sont plus élevées, souvent le double de celles utilisées dans le SQTL.
La posologie est augmentée jusqu’à obtention de la suppression des phénomènes répétitifs ; le tout doit être vérifié par une épreuve d’effort de contrôle sous traitement vérifiant l’absence de phénomène répétitif grave à l’effort.
Néanmoins, il est probable que l’indication de défibrillateur implantable chez l’adolescent ou le jeune adulte des TVC incomplètement contrôlées par une posologie de bêtabloquants pourrait être envisagée plus largement dans l’avenir.
Bilan d'une série française
Nous avons rapporté récemment une série de plus de 100 patients, dont 50 propositus et 51 membres des mêmes familles avec un suivi de 8 ans. Dans cette série rétrospective, 60 % des patients étaient symptomatiques avant le diagnostic, en particulier tous les probants. Les patients étaient symptomatiques avant l’âge de 20 ans dans 93 % des cas, tous étaient symptomatiques à l’exercice. Le délai entre la 1re syncope et le diagnostic est de 2 ans et demi, d’où l’importance de faire un bilan le plus complet possible d’une syncope d’effort chez l’enfant ou l’adolescent. Les facteurs de risque d’événement cardiaque au diagnostic sont l’âge jeune au diagnostic, l’absence de bêtabloquants. Les facteurs de risque d’événement cardiaque durant le suivi sont l’absence de bêtabloquants, une histoire de mort subite familiale ou d’antécédent de mort subite. Dernier élément important de cette série française, l’absence de différence entre les propositus et les membres de la famille en ce qui concerne la survenue d’événement cardiaque. L’enquête génétique prend là toute son importante et la prise en charge qui en découle implique une surveillance rapprochée et un traitement, même chez les sujets asymptomatiques ayant la mutation.
En pratique
Une syncope d’effort chez un enfant ou un adolescent est toujours un événement grave qui nécessite un bilan complet à la recherche des causes familiales de mort subite que sont la cardiomyopathie hypertrophique, la cardiomyopathie dilatée, les syndromes de Marfan, éventuellement la dysplasie arythmogène du ventricule droit, mais surtout les canalopathies en particulier le SQTL dans la forme LQT1 ou LQT2 et les TVC.
Ces maladies familiales, dont 50 % de collatéraux et descendants peuvent être touchés, impliquent une enquête génétique multidisciplinaire pour proposer aux familles un diagnostic étiologique et une prise en charge adaptée. La découverte d’une canalopathie de type SQTL ou TVC implique l’interdiction de la compétition, l’autorisation de certains sports adaptés suivant les recommandations des sociétés savantes (www.cardiogen.fr). Une activité de loisir peut être discutée chez les sujets ayant une forme asymptomatique de SQTL traitée par bêtabloquants. Des loisirs sont encore plus restreints pour les TVC asymptomatiques traités par bêtabloquants et les sujets asymptomatiques ayant une cardiomyopathie hypertrophique ayant moins de dix ans.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :

Articles sur le même thème
publicité
publicité