




Publié le 08 nov 2011Lecture 10 min
Cardiopathies amyloïdes : nouvelles données
T. DAMY, S. GUENDOUZ, K. BELHADJ, J.-F. DEUX, N. BENHAIEM, V. AUDARD, M. GOOSSENS, D. AZOULAY, J.-P. COUETIL, V. PLANTE-BORDENEUVE, Réseau Amylose Mondorien, CHU Henri-Mondor, Créteil
Les Journées françaises de l'insuffisance cardiaque
Depuis dix ans la prise en charge de l’amylose a beaucoup progressé tant en termes de diagnostic étiologique, de traitement que de pronostic.
Le cardiologue est souvent à l’origine du diagnostic d’amylose cardiaque lors de la réalisation d’une échocardiographie pour un bilan de dyspnée, lors de bilan de trouble de la conduction ou lors de la découverte d’un rehaussement tardif à l’IRM cardiaque après injection de gadolinium. On ne peut plus conclure en 2011 qu’un patient avec une forme cardiaque d’amylose est condamné à 6 mois. Il est nécessaire de prendre en considération les nouvelles données.
Définition et classification des amyloses
L’amylose est la manifestation de plusieurs maladies systémiques. La caractéristique commune de ces maladies est une accumulation extracellulaire de protéines fibrillaires insolubles qui se déposent et envahissent progressivement les tissus, empêchant leur bon fonctionnement. Ces protéines, quelle que soit leur origine, ont la particularité de fixer le rouge Congo en coupe d’anatomopathologie et d’avoir une biréfringence (jaune-verte) en lumière polarisée.
La classification des amyloses repose sur la nature biochimique de la protéine amyloïde impliquée dans la formation des dépôts. Les formes les plus fréquentes sont les amyloses AL (immunoglobuliniques) et les amyloses à transthyrétine (TTR), héréditaire (TTR mutée) et sénile (TTR sauvage) (tableau).
Les amyloses AL
Elles sont liées principalement aux gammapathies monoclonales et au myélome. Les gammapathies monoclonales sont très fréquentes et touchent environ 10 % des patients de 60 ans. Heureusement, ces gammapathies se compliquent rarement d’amylose mais représentent plus de 60 % des cardiopathies amyloïdes.
Les amyloses à transthyrétine
La transthyrétine (TTR) est une protéine synthétisée par le foie sous forme de monomère. Ces monomères s’assemblent en tétramères qui transportent des protéines (ex : hormone thyroïdienne) dans le sang. Les amyloses TTR sont de deux types :
• La forme familiale où la transthyrétine est mutée (ATTRm). La transmission est autosomique dominante. Plus de 100 mutations pathogènes du gène codant pour la TTR ont été identifiées entraînant une déstabilisation du tétramère qui se dissocie en monomères à l’origine de l’accumulation tissulaire de fibrilles. Les dépôts tissulaires comportent également de la transthyrétine non mutée. Bien qu’héréditaire, il arrive fréquemment dans les cas « sporadiques » d’âge de début tardif que l’on ne retrouve pas d’antécédents familiaux.
• La forme sénile où le précurseur est la transthyrétine non mutée (ATTRwt) survient quasi-exclusivement chez des hommes âgés. Dans ces cas, c’est la dissociation du tétramère « sauvage » qui est à l’origine des dépôts amyloïdes, probablement sous l’influence de facteurs liés au vieillissement.
Les amyloses AA
L’amylose AA est caractérisée par la présence de dépôts intratissulaires de fibrilles constituées de fragments N terminaux autoagrégés de la protéine amyloïde A (qui dérive du clivage de la serum amyloid A protein, SAA). La protéine SAA est une apolipoprotéine synthétisée au niveau du foie, dont la concentration sérique augmente considérablement au cours des maladies inflammatoires chroniques (rhumatismales, digestives, fièvres héréditaires familiales). L’atteinte rénale est le plus souvent au premier plan mais des cardiopathies amyloïdes ont aussi été décrites chez des patients présentant une amylose AA.
Manifestations tissulaires
Les fibrilles amyloïdes peuvent se déposer dans tous les tissus. Les manifestations cliniques apparaissent à des âges variables, des degrés variables et des localisations tissulaires différentes suivant le type d’amylose. Dans les amyloses AL, les dépôts sont ubiquitaires. Dans les amyloses héréditaires à transthyrétine, la mutation « Val30Met » est la plus fréquente et se traduit chez les patients d’origine portugaise par une atteinte neurologique précoce (30 ans) et une atteinte cardiaque plus tardive. Dans les formes sporadiques françaises, l’atteinte peut-être mixte (cardiaque et neurologique) ou uniquement cardiologique (Val122Ile ; Ser77Tyr) et survient généralement chez des patients plus âgés (60 ans). Les amyloses séniles sont uniquement cardiaques.
Les atteintes cardiaques
Les manifestations cardiaques sont liées aux dépôts amyloïdes au sein du tissu interstitiel et des structures vasculaires et nerveuses myocardiques. Ces atteintes peuvent être associées ou dissociées. C’est-à-dire qu’un patient peut avoir une « dénervation » cardiaque sans hypertrophie ou avec hypertrophie minime, notamment dans le cas des amyloses héréditaires portugaises (mutationVal30Met).
Lorsqu’il y a des dépôts interstitiels myocardiques, ceux-ci vont induire un épaississement de l’ensemble du tissu myocardique expliquant la « fausse » hypertrophie des deux ventricules évoluant vers une altération de la fonction diastolique (restriction) et secondairement de la fonction systolique.
Ces atteintes se traduisent au plan clinique par des symptômes d’insuffisance cardiaque (dyspnée, etc.) et des troubles rythmiques fréquents à l’étage auriculaire (flutter et FA) et moins fréquents à l’état ventriculaire.
L’ECG retrouve un microvoltage. Il n’y a pas d’hypertrophie cardiaque à l’ECG, puisque les dépôts sont interstitiels et que la taille des cardiomyocytes n’est pas augmentée.
L’échocardiographie va permettre la quantification de l’épaisseur des parois, et l’évaluation d’un épaississement du septum interauriculaire et des valves. L’aspect scintillant, classiquement associé aux dépôts amyloïdes, est non spécifique. Le profil transmitral peut être restrictif (E/A > 2). Enfin, on observe fréquemment un épanchement péricardique qui peut être abondant et, dans certains cas, évolue vers la tamponnade.
Le diagnostic est très évocateur lorsque s’associent un microvoltage à l’ECG, une hypertrophie biventriculaire et un épanchement péricardique à l’échocardiographie (figure 1).
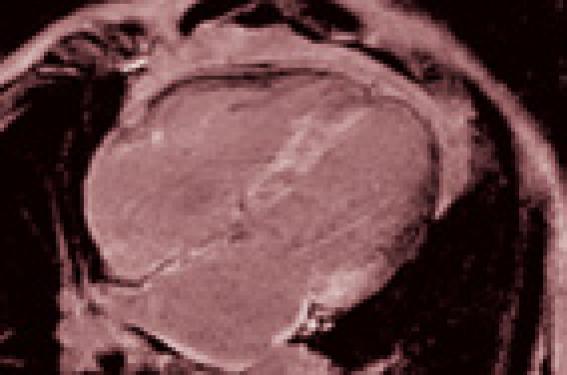
L’IRM retrouve les modifications structurelles objectivées à l’échocardiographie et a l’intérêt de mettre en évidence les dépôts amyloïdes sous forme d’un rehaussement tardif après injection de gadolinium (figure 2). Ces dépôts sont diffus et sous-endocardiques dans les formes sévères détectées tardivement, mais peuvent être plus limités dans les formes vues précocement.
Figure 1. Échocardiographie d’une amylose cardiaque typique.
a) Vue parasternale grand axe : amylose à transthyrétine (Val30Met) chez une patiente de 62 ans : hypertrophie biventriculaire et épanchement péricardique. b) Vue parasternale grand axe : amylose AL (chaîne lambda) chez une patiente de 35 ans ; hypertrophie importante du ventricule gauche. c) Vue 4 cavités : amylose à transthyrétine (Ser77tyr) chez un patient de 78 ans. Hypertrophie biventriculaire, dilatation des oreillettes, épaississement des feuillets valvulaires et du septum interauriculaire avec IT importante.
Figure 2. IRM cardiaque : rehaussement tardif diffus (dépôts blancs) au sein de toutes les parois du cœur (VG, VD, OG, OD, SIA). Amylose à transthyrétine.
Les dépôts au sein du tissu cardionecteur vont être responsables des troubles de la conduction à type de bloc auriculo-ventriculaire. Ils se traduisent cliniquement par l’apparition de malaise et de syncope. Ces blocs sont d’apparition progressive ou favorisés par l’administration de traitement médical dromotrope négatif. Les morts subites sont fréquentes dans l’amylose. Leur origine est discutée suivant les types d’amylose et les arythmies ventriculaires semblent nettement moins fréquentes que les dissociations auriculo-ventriculaires. Ces dernières pourraient être favorisées par le stress.
Les atteintes extracardiaques
Les atteintes neurologiques prédominent sur le système nerveux autonome et sur les nerfs périphériques avec une atteinte des fibres longueurs-dépendantes.
L’atteinte nerveuse périphérique se traduit cliniquement par des troubles sensitifs associés à des paresthésies des extrémités. L’atteinte du système nerveux autonome peut être au premier plan et toucher toutes les fonctions autonomes entraînant des gastroparésies responsables de vomissements incoercibles, sources de dyskaliémie, de troubles des fonctions génito-urinaires, responsables d’infection, et d’hypotensions orthostatiques sévères. Les autres tissus touchés sont le rein (protéinurie, syndrome néphrotique), la langue (macroglossie), l’estomac (gastrite), les yeux (dépôts vitréens, cornéens, glaucome et hématome péri-orbitaire) et les canaux carpiens. Un amaigrissement, parfois très important, est observé dans toutes les formes. Il n’est malheureusement pas rare, devant la multiplicité des symptômes, que le patient ne soit pas pris au sérieux et que le diagnostic traîne.
Diagnostic des amyloses
Les examens cités ci-dessus vont permettre de suspecter l’amylose. L’origine AL va être recherchée par la réalisation d’une électrophorèse et d’une immunofixation des protides sanguins et urinaires. Ces examens seront complétés par le dosage des chaînes libres Kappa et Lambda réalisé par néphélémétrie. En cas de gammapathie, la production d’une chaîne sera prédominante et le ratio des chaînes Kappa/Lambda déséquilibré. Cet examen permettra également d’évaluer la réponse à la chimiothérapie. Il est à souligner, d’après deux études, qu’environ 5 % des formes supposées AL sont en fait des amyloses héréditaires. Ceci s’explique par la fréquence des gammapathies, notamment dans la population âgée de plus de 65 ans.
L’anatomopathologie permet d’authentifier les dépôts d’amylose par biopsies. Cette analyse peut être faite en première intention à partir de biopsie de la graisse péri-ombilicale ou des glandes salivaires, qui sont les tissus les plus accessibles. La négativité de ces biopsies ne permet pas d’éliminer le diagnostic puisque la distribution des dépôts amyloïdes est patchy.
La réalisation de biopsies plus invasives, myocardique, rénale ou nerveuse, est donc souvent nécessaire. Les colorations comportent le rouge Congo et la Thioflavine T. Le rouge Congo est examiné en lumière polarisée et donne une biréfringence jaune-vert. L’immunofixation avec des anticorps marqués dirigés contre les protéines recherchées (chaîne lambda, kappa, transthyrétine, AA, etc.) pourra aider à caractériser la nature du dépôt amyloïde (figure 2). Cependant, l’immunomarquage peut être non spécifique. Le recours à des techniques sophistiquées comme la spectrométrie de masse est parfois nécessaire.
Le diagnostic des formes héréditaires repose sur les tests génétiques avec séquençage du gène ATTR de la transthyrétine.
L’amylose sénile de localisation cardiaque reste finalement un diagnostic d’élimination. Il est souvent difficile éthiquement d’aller jusqu’à la biopsie myocardique pour affirmer le diagnostic chez un sujet âgé en l’absence de traitement spécifique.
Figure 3. Anatomopathologie de l’amylose. a) Biopsie myocardique : coloration haemalin-éosine retrouvant des dépôts d’amylose très importants aboutissant à une déstructuration complète du tissu myocardique et à la disparition d’une grande partie des cardiomyocytes. b) Biopsie myocardique : immunomarquage transthyrétine positive. Les dépôts de transthyrétine apparaissent de couleur marron. Noter l’importance des dépôts qui entourent complètement les cardiomyocytes.
Les traitements spécifiques des amyloses
De nouvelles molécules (chimiothérapies) sont apparues dans l’amylose AL (dexaméthasone-melphalan et plus récemment bortézomib (Velcade®). Elles permettent d’observer des taux de rémission (normalisation des chaînes légères dans le sang) plus fréquents que par le passé. Secondairement, apparaît une stabilisation et/ou une régression des atteintes organiques se traduisant au niveau cardiaque par une diminution du NT-proBNP et de la troponine puis une régression du microvoltage.
Ces molécules ont permis une amélioration considérable du pronostic des patients.
De nouvelles chimiothérapies et protocoles sont en cours d’étude.
Dans l’amylose à transthyrétine, la greffe hépatique est utilisée depuis plus de 10 ans. Elle permet de stabiliser l’atteinte neurologique à condition d’être réalisée à un stade précoce de l’évolution. Son effet sur l’atteinte cardiaque est moins net. De nouveaux traitements sont développés dans le cadre de protocoles de recherche. Une molécule augmentant la stabilisation de la transthyrétine pourrait ralentir l’atteinte neurologique.
Points à retenir
- Le pronostic des amyloses a évolué avec l’apparition de nouveaux traitements.
- Le cardiologue est en première position pour évoquer le diagnostic d’amylose cardiaque grâce à l’échocardiographie et à l’IRM.
- Il est nécessaire de rechercher la cause de l’amylose afin de mettre en route le traitement approprié.
- La prise en charge des patients dans les centres spécialisés est nécessaire.
Traitements des cardiopathies amyloïdes
Il existe peu ou pas d’étude sur le traitement des cardiopathies amyloïdes.
En cas de symptôme d’insuffisance cardiaque, il est recommandé d’utiliser un diurétique de l’anse plus ou moins associé à de l’aldactone.
Les bêtabloquants et inhibiteurs de l’enzyme conversion, recommandés classiquement chez l’insuffisant cardiaque, peuvent avoir des effets secondaires graves chez les patients amyloïdes en diminuant le débit cardiaque, majorant les troubles de la conduction et la dysautonomie (hypotension orthostatique +++).
La digoxine n’est pas recommandée en raison d’un risque de toxicité tissulaire du fait de son accumulation possible sur les fibrilles amyloïdes.
Le traitement anticoagulant doit être discuté en cas de cardiopathie. Les cardiopathies amyloïdes présentent fréquemment des thrombi intracardiaques qui se compliquent d’accidents emboliques.
L’indication d’implantation des pacemakers doit être large chez ces patients du fait des troubles de la conduction.
Ces derniers doivent être dépistés par des Holters et aussi par des explorations électrophysiologiques. Une surveillance rythmologique s’impose. L’implantation de défibrillateur implantable pour prévenir la mort subite est controversée et doit être discutée au cas par cas.
La greffe cardiaque est envisageable et a été réalisée dans tous les types d’amylose (amylose AL comprises). Devant la complexité de ces pathologies, il est préférable de discuter de cette indication dans des centres multidisciplinaires spécialisés. Dans le cadre des amyloses à transthyrétine, la greffe cardiaque devra être associée simultanément ou de manière différée à une greffe hépatique pour éviter la récidive de l’amylose sur le greffon.
En pratique
De nouvelles avancées ont été réalisées dans le diagnostic et le traitement des amyloses. Le pronostic des cardiopathies amyloïdes est bien meilleur qu’il ne l’était il y a 10 ans.
Il est nécessaire que ces patients bénéficient d’une prise en charge adaptée et spécialisée, afin que le diagnostic étiologique puisse être assuré et la thérapeutique adaptée.
Le cardiologue a une position centrale puisqu’il dépiste fréquemment de nouveaux cas et doit assurer la prise en charge et le dépistage des atteintes cardiaques.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :

Articles sur le même thème
publicité
publicité