

Publié le 18 sep 2007Lecture 13 min
Diabète et endothélium : la dysfonction endothéliale du diabétique ; physiopathologie, explorations, qu'en conclure ?
A. NITENBERG, CHU Jean Verdier, Bondy
L’athérosclérose est une cause majeure de la morbidité et de la mortalité des diabétiques. L’endothélium, qui participe au contrôle de nombreuses fonctions vasculaires, est une cible privilégiée du diabète : il intervient dans la modulation de la vasomotricité, la coagulation, les mécanismes d’adhésion et de migration cellulaires, les processus inflammatoires, les mécanismes de remodelage vasculaire, la perméabilité de la paroi, et l'induction et le développement de la plaque d'athérome. Le stress oxydant et la diminution de la biodisponibilité du monoxyde d’azote sont des étapes clés de la dysfonction endothéliale du diabétique, dont une des manifestations est la dépression de la vasodilatation endothélium-dépendante. L’hyperglycémie étant un stimulus direct du stress oxydant, le contrôle strict de la glycémie reste le meilleur garant de l’amélioration de la fonction endothéliale et de la prévention de l’athérosclérose et des événements cardiovasculaires chez les diabétiques.
Le diabète est une maladie et un facteur de risque vasculaire indépendant qui favorise le développement précoce de l'athérosclérose. Les maladies cardiovasculaires, et en premier lieu les cardiopathies ischémiques, constituent la première cause de la morbidité et de la mortalité chez les patients diabétiques quels que soient les autres facteurs de risque, 70 à 80 % des décès des diabétiques étant liés à une complication vasculaire. Chez ces patients, la maladie coronaire est de 2 à 4 fois plus fréquente, les accidents vasculaires cérébraux de 1,4 à 2,2 fois plus fréquents, l’artériopathie périphérique plus de 10 fois plus fréquente que dans la population non diabétique.
Des anomalies vasculaires sont identifiables bien avant le développement de l’athérosclérose et il est maintenant reconnu que ces anomalies sont liées à une dysfonction de l’endothélium vasculaire.
L’endothélium, qui participe au contrôle de nombreuses fonctions vasculaires, intervient dans :
- la modulation de la vasomotricité,
- la coagulation,
- les mécanismes d’adhésion et de migration cellulaires,
- les processus inflammatoires,
- la perméabilité vasculaire,
- les mécanismes de remodelage vasculaire,
- l'induction et le développement de la plaque d'athérome.
Physiopathologie de la dysfonction endothéliale du diabétique
Le stress oxydant, un élément-clé de la dysfonction endothéliale du diabétique
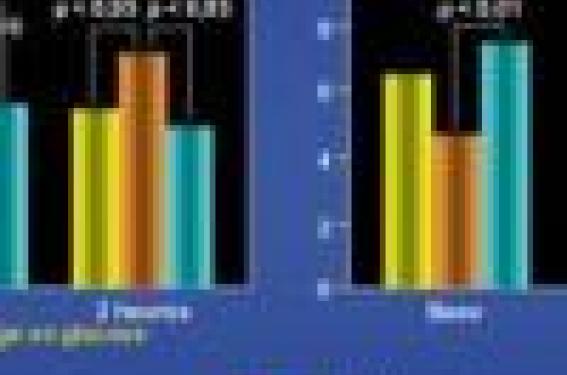
L’augmentation de la production d’anions superoxydes est un fait avéré dans le diabète. Ces anions superoxydes sont produits en excès par les cellules endothéliales, mais également par les monocytes et les cellules musculaires lisses. Chez l’homme, la concentration en acide thiobarbiturique (TBAR), un marqueur du stress oxydant, est augmentée chez les diabétiques de type 2, chez les sujets insulinorésistants et après une charge en glucose chez les sujets normaux, ce qui démontre le rôle indépendant de la glycémie (figure 1). Cette production accrue d’anions superoxydes a plusieurs origines :
- les NADH/NAD(P)H oxydases membranaires stimulées par l’hyperglycémie, via l’aldose réductase et la protéine kinase C (figure 2) ;
- le stress réducteur lié à l’oxydation du glucose qui stimule la xanthine oxydase mitochondriale en même temps qu’il diminue l’action protectrice de la superoxyde dismutase et de la glutathion réductase ;
- les produits de la glycation (AGE) qui stimulent des récepteurs cellulaires spécifiques (RAGE) ;
- l’augmentation d’électrons libres, qui se combinent avec l’oxygène, due à la diminution de la fonction accepteur d’électrons de la NO-synthase.
Figure 1. À gauche, une charge en glucose augmente la concentration en acide thiobarbiturique (TBAR) dans le sang aussi bien chez les sujets diabétiques de type 2 ou intolérants au glucose que chez les sujets normaux. À droite, on observe une dépression de la dilatation flux-dépendante parallèle à l’augmentation du stress oxydant, y compris chez les sujets normaux mais à un moindre degré que chez les sujets diabétiques de type 2 ou intolérants au glucose. (d’après Kawano et coll. J Am Coll Cardiol 1999 ; 34 : 146-54).
Figure 2. Le diabète est à l’origine d’une cascade d’événements qui stimulent le stress oxydant. Une des voies de stimulation du stress oxydant passe par les NAD(P)H oxydases membranaires. L’augmentation des radicaux superoxydes active le facteur nucléaire NF-kB et inactive le monoxyde d’azote (NO).
Cette stimulation du stress oxydant chez le diabétique est à l’origine d’une cascade d’événements qui résultent de la dépression de la production de monoxyde d’azote et de l’activation du nuclear factor-kB (NF-kB) par les cellules endothéliales.
La dépression de biodisponibilité du monoxyde d’azote (NO)
La biodisponibilité du monoxyde d’azote peut être diminuée de deux manières par le stress oxydant, soit par diminution de la synthèse, soit par augmentation de l’inactivation.
Diminution de la production de monoxyde d’azote
Bien que, chez le sujet normal, l’insuline stimule la production de monoxyde d'azote par l'endothélium, la synthèse du monoxyde d'azote pourrait être diminuée en raison d'un déficit d'apport en substrat de la NO-synthase, la L-arginine, mais les résultats sont contradictoires. Cependant, dans l’hyperinsulinisme du diabète de type 2, le déficit de production de monoxyde d’azote pourrait être consécutif à l’oxydation de la tétrahydrobioptérine, cofacteur de la NO-synthase par le stress oxydant, ou résulter de l’augmentation de la diméthylarginine asymétrique chez les diabétiques, un antagoniste de la L-arginine.
Inactivation du monoxyde d’azote
L’augmentation de la production de radicaux superoxydes, qui ont une grande affinité pour le monoxyde d’azote – dont la durée de vie est spontanément très courte (0,5 à 1 s) –, conduit à l’inactivation de ce dernier et sa transformation en peroxynitrites (ONOO-) très toxiques pour les membranes cellulaires (figure 3).
Figure 3. La combinaison du monoxyde d’azote (NO) et des radicaux superoxydes conduit à la formation de péroxynitrites (ONOO-) très toxiques pour les membranes cellulaires. La diminution de la biodisponibilité du NO est responsable d’une dépression de la relaxation des cellules musculaires lisses et de la vasodilatation endothélium-dépendante.
L’activation du NF-kB
Le NF-kB contrôle une multitude de gènes et a donc des effets nucléaires pléiotropes (figure 4) ; il stimule :
- la production des cytokines pro-inflammatoires (IL2, IL6, IL8, TNFa), qui en retour stimulent la protéine C réactive (CRP) ;
- les molécules d’adhésion cellulaires (ICAM1, VCAM1 et E-sélectine), qui activent l’adhésion des monocytes et des lymphocytes T sur l’endothélium ;
- les facteurs de migration cellulaire (MCP1) dans l’intima des vaisseaux ;
- l’activation de la cyclooxygénase 2 (COX2) et la production de prostaglandine H2 vasoconstrictrice ;
- surtout, la synthèse de l’angiotensinogène cellulaire (et la production d’angiotensine II endogène qui en résulte), et de l’ARN messager des récepteurs AT1, ce qui accroît le nombre des récepteurs membranaires. Cette activation du système rénine-angiotensine stimule le système orthosympathique, augmente en retour le stress oxydant en agissant sur les NADH/NADPH membranaires. Elle stimule également la contraction, l’hypertrophie et l’hyperplasie des cellules musculaires lisses vasculaires par l’intermédiaire des mitogen activator protein kinases (MAPK), la production d’endothéline par les cellules endothéliales (cette dernière étant elle-même vasoconstrictrice et hypertrophiante de la paroi vasculaire), les métalloprotéinases – facteurs de dégradation de la trame collagène –, favorisant le remodelage de la paroi vasculaire, la fibrose interstitielle et périvasculaire par l’intermédiaire de l’aldostérone, l’endocytose du LDL oxydé par les cellules endothéliales et les macrophages liée à l’augmentation de l’expression des récepteurs du LDL oxydé LOX1 et CD36.
Figure 4. Représentation schématique de la cascade d’événements consécutive au diabète et à l’augmentation du stress oxydant (.O2-) au niveau de la cellule endothéliale. ARNm : ARN messager ; AT1R : récepteur de l’angiotensine II ; CRP : protéine C réactive ; ET-1 : endothéline-1 ; LOX1 : récepteur du LDL oxydé ; MAPK : mitogen activator protein kinase ; NF-kB : nuclear factor-kB ; S : système orthosympathique.
Exploration de la dysfonction endothéliale chez les diabétiques
Parce que historiquement la dépression de la vasodilatation endothélium-dépendante a été la première manifestation décrite de la dysfonction endothéliale, l’exploration de la vasodilatation endothélium-dépendante est un des moyens les plus simples de mettre en évidence la dysfonction endothéliale. En effet, l’intégrité de la fonction endothéliale se traduit par une dilatation des vaisseaux de conductance et de la microcirculation en réponse à des stimuli qui augmentent la production de monoxyde d’azote (NO) par les cellules endothéliales.
Cette augmentation de la synthèse de NO – et la vasodilatation consécutive – peut être induite soit par des agents pharmacologiques qui agissent sur des récepteurs spécifiques de la cellule endothéliale (acétylcholine ou analogue, sérotonine, adénosine diphosphate, substance P), soit par un facteur mécanique tel que la vitesse d’écoulement du sang (contrainte de cisaillement longitudinale) qui peut être augmentée par l’administration d’un vasodilatateur endothélium-indépendant ou par l’augmentation du métabolisme en aval du site de mesure, ce qui augmente le débit au niveau du site de mesure (dilatation flux-dépendante). Une dysfonction endothéliale est avérée lorsque la dilatation est simplement diminuée ou remplacée par une vasoconstriction.
Des anomalies de la vasodilatation endothélium-dépendante ont été mises en évidence chez les diabétiques dans tous les territoires vasculaires. Ces anomalies sont observées aussi bien sur les troncs artériels, que sur la microcirculation. Pour les troncs artériels, elle se traduit par une absence d’augmentation ou une diminution de calibre, pour la microcirculation, elle se traduit par une absence d’augmentation ou une diminution de débit.
Dysfonction endothéliale des troncs artériels chez les diabétiques
L’injection d’acétylcholine, induit une vasoconstriction chez les diabétiques de type 1 et 2 (figure 5). Chez les diabétiques de type 2, la vasodilatation flux-dépendante des artères de l’avant-bras et des artères coronaires angiographiquement normales est diminuée ou totalement abolie. La responsabilité de l’endothélium est avérée par le fait que le calibre luminal des artères est comparable à celui de sujets témoins, tant à l'état basal qu'après vasodilatation maximale par un dérivé nitré.
Figure 5. À gauche, chez des patients sans autre facteur de risque coronaire que le diabète et présentant des artères coronaires angiographiquement normales, l’injection intracoronaire d’acétylcholine induit une vasoconstriction dose-dépendante alors que chez les sujets témoins, on observe une dilatation dose-dépendante. On observe également que la vasodilatation endothélium-indépendante au dinitrate d’isosorbide (DNI) est préservée chez les diabétiques (D’après Nitenberg et coll. Diabetes 1993 ; 42 : 1017-25). À droite, la dilatation de la microcirculation coronaire estimée par l’augmentation du débit coronaire est également déprimée chez les diabétiques, comme en atteste l’absence d’augmentation du débit coronaire lors d’un test au froid (D’après Nitenberg et coll. Diabetes 2001; 50 : 80-5).
Dysfonction endothéliale des artérioles chez les diabétiques
L’injection de métacholine, un analogue de l’acétylcholine, dans une artère de l’avant-bras, induit chez les diabétiques de type 2 une augmentation du débit inférieure à celle de sujets normaux et il y a une dépression de l’augmentation du débit lors de l’hyperémie postocclusive chez les diabétiques de type 1.
Des anomalies de la vasomotricité microcirculatoire ont également été montrées dans d’autres territoires tels que la circulation cérébrale ou la circulation coronaire, où l’augmentation de la demande métabolique du myocarde induite par un test au froid (stimulation sympathique) provoque une augmentation du débit coronaire beaucoup plus faible chez les diabétiques que chez les sujets normaux sans corrélation avec la demande métabolique du myocarde (figure 5).
De même, l’augmentation de la fréquence cardiaque entraîne une réduction de la résistance coronaire significativement moins importante chez les sujets diabétiques. Il s’agit, là aussi, d’anomalies fonctionnelles comme en atteste l'absence d'adaptation du débit coronaire à la demande métabolique du myocarde alors que le débit coronaire pourrait être potentiellement augmenté puisque les valeurs mesurées lors du test au froid ou lors de l'augmentation de la fréquence cardiaque sont loin d'atteindre les valeurs de débit maximal mesurées après vasodilatation maximale de la microcirculation.
Conséquences de la dysfonction endothéliale
Le stress oxydant et la dysfonction endothéliale consécutive représentent une explication plausible de l’ensemble des complications vasculaires du diabète. En effet, la dysfonction endothéliale a des conséquences vasculaires multiples.
Les anomalies de la vasomotricité
Malgré quelques résultats discordants sur la vasomotricité des artères de l'avant-bras, il semble bien que la diminution de la biodisponibilité du NO consécutive au stress oxydant soit responsable des anomalies de la vasodilatation endothélium-dépendantes chez les diabétiques. Cette hypothèse est soutenue par des arguments directs et indirects :
- chez l’homme, l’augmentation de la concentration en TBAR chez les diabétiques de type 2, chez les sujets insulinorésistants et après une charge en glucose chez les sujets normaux, est corrélée à la dépression de la vasodilatation endothélium-dépendante de l’artère brachiale (figure 1) ;
- l’augmentation de la glycémie après les repas est responsable d’une augmentation de la concentration sanguine en nitrotyrosine (un autre marqueur du stress oxydant) et d’une dépression de la vasodilatation endothélium-dépendante ;
- les agents antioxydants rétablissent la relaxation endothélium-dépendante. Ainsi chez l'homme, l'administration de vitamine C rétablit une vasomotricité normale au niveau de l'avant-bras ; l’administration de déferoxamine, un inhibiteur de la production de radicaux hydroxyles, rétablit une réponse vasodilatatrice normale des artères coronaires épicardiques et de la microcirculation coronaire au test au froid et à l'augmentation de la vitesse d'écoulement intracoronaire.
Il convient par ailleurs de signaler que l’existence d’une dépression de la vasodilatation endothélium-dépendante est un facteur prédictif indépendant d’événement coronaire chez le diabétique (figure 6).
Figure 6. Chez les sujets diabétiques, l’existence d’une constriction coronaire lors du test au froid (groupe 2) est prédictive de la survenue d’événements coronaires alors que, chez les sujets ne présentant pas de constriction (groupe 1), la fréquence des événements est comparable à celle des sujets témoins (D’après Nitenberg et coll. Diabetes Care 2004 ; 27 : 208-15).
L’athérosclérose
Il est intéressant de noter que les radicaux libres de l'oxygène, qui sont fortement impliqués dans la genèse des anomalies de la vasomotricité endothélium-dépendante, sont également responsables de l'oxydation du LDL-cholestérol, qui est une étape indispensable à l'accumulation de celui-ci dans les macrophages. En effet, et bien que le développement de l'athérosclérose coronaire chez les diabétiques soit multifactoriel, dans tous les cas l'endothélium vasculaire y joue un rôle déterminant par les radicaux libres de l'oxygène produits en grande quantité et l’oxydation du LDL-cholestérol qui en résulte (figure 7).
Figure 7. Le stress oxydant consécutif au diabète est un puissant inducteur de l’athérosclérose car, en même temps qu’il active le facteur nucléaire NF-kB, il oxyde le LDL-cholestérol, une étape déterminante dans l’accumulation du cholestérol par les monocytes qui ont migré dans l’intima via la stimulation des molécules d’adhésion (ICAM1, VCAM1, P-sélectine) et le facteur de migration (MCP1). Transformés en macrophages, ils sont également activés, en particulier par l’interféron g (INFg) produit par les lymphocytes T, et forment les cellules spumeuses à l’origine de la plaque d’athérome.
Chez un sujet normal, la captation du LDL-cholestérol par les macrophages, qui dépend de récepteurs spécifiques de la membrane, est limitée par un mécanisme de rétrocontrôle cellulaire négatif qui n'existe plus lorsque le LDL-cholestérol est oxydé. Les macrophages qui ont migré dans l’intima et dont l’expression des récepteurs LOX1 et CD36 est augmentée par le diabète, accumulent alors le LDL-cholestérol oxydé pour former les cellules spumeuses et ce, jusqu'à la mort cellulaire, initiant ainsi le cercle vicieux conduisant à la formation de la plaque d'athérome. Enfin, le LDL-cholestérol ainsi oxydé est lui-même un facteur de dysfonction endothéliale, car il inactive également le monoxyde d'azote, et active le NF-kB (figure 4).
Les anomalies de la coagulation et la thrombose
La diminution de la biodisponibilité du monoxyde d’azote facilite l’agrégation plaquettaire, le diabète favorise la coagulation en diminuant l’héparan sulfate, en augmentant celle de PAI1 et de facteur von Willebrand, et déprime la fibrinolyse en diminuant la production endothéliale de t-PA.
Au total, les altérations fonctionnelles et anatomiques de la paroi vasculaire, l’activation plaquettaire et les anomalies de l’hémostase constituent donc un terrain extrêmement propice à la rupture de plaque et à la thrombose.
La perméabilité vasculaire
L’augmentation de la perméabilité de l’endothélium dans le diabète, facilite le transfert du LDL-cholestérol vers l’intima où il est oxydé. Par ailleurs, il est suggéré que l’existence d’une microalbumiurie est le témoin d’un dysfonctionnement généralisé de l’endothélium, sans toutefois que cela soit formellement établi et bien qu’une corrélation ait été retrouvée entre microalbuminurie et dépression de la vasodilatation endothélium-dépendante chez le diabétique (figure 8).
Figure 8. À gauche, les sujets diabétiques de type 2 ayant une microalbuminurie ont une vasoconstriction coronaire plus marquée que les sujets normoalbuminuriques lors du test au froid. À droite, il existe une corrélation négative entre l’excrétion urinaire d’albumine urinaire et la diminution du diamètre coronaire lors du test au froid (D’après Cosson et coll. Diabetes Care 2006 ; 29 : 107-12).
En pratique
Bien que l’augmentation du stress oxydant soit un élément- clé de la dysfonction endothéliale dans le diabète, les antioxydants qui inactivent les radicaux libres de l’oxygène, s’ils améliorent la fonction endothéliale à très court terme chez l’homme, n’ont pas d’efficacité à long terme. Cela ne signifie pas pour autant que le stress oxydant n’est pas impliqué dans les complications cardiovasculaires du diabète, mais nous ne disposons pas à l’heure actuelle de moyens adaptés pour combattre la production de radicaux libres de l’oxygène.
Cependant, l’effet bénéfique des inhibiteurs de l’enzyme de conversion mis en évidence dans de nombreuses études, qui montrent une réduction significative de la morbidité et de la mortalité cardiovasculaires, pourrait en partie être lié à une diminution de la production NAD(P)H-dépendante de radicaux superoxydes. Une part des effets bénéfiques des statines pourrait également être liée à un effet antioxydant, mais aucune preuve directe démontrant que ces effets bénéfiques soient dus à un effet antioxydant n’a été apportée.
In fine, l’hyperglycémie à elle seule étant un stimulus direct du stress oxydant, le contrôle strict de la glycémie doit rester le premier objectif thérapeutique puisque celui-ci est un des meilleurs garants de l’amélioration de la fonction endothéliale et probablement de la prévention des événements cardiovasculaires chez les diabétiques. En effet, même si l’adjonction d’un antagoniste du système rénine-angiotensine et d’une statine a prouvé son efficacité, celle-ci est conditionnée par le contrôle du diabète.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :

Articles sur le même thème
publicité
publicité